

 產(chǎn)業(yè)資訊
產(chǎn)業(yè)資訊
 醫(yī)藥時間
醫(yī)藥時間  2024-06-06
2024-06-06
 369
369
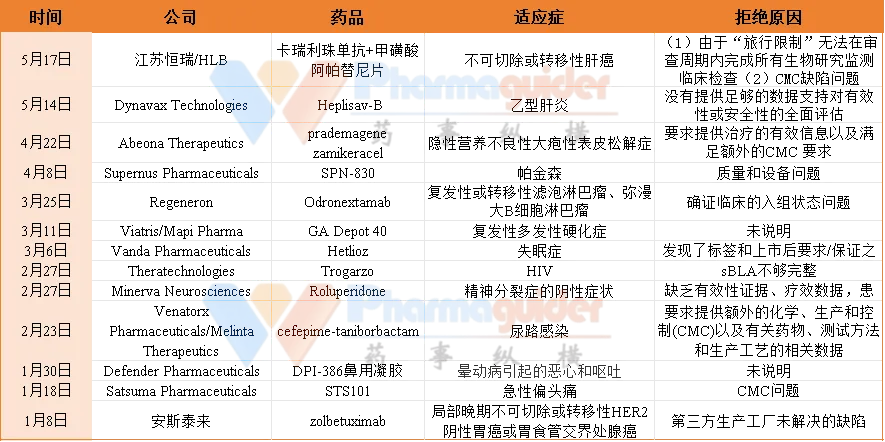
據(jù)不完全統(tǒng)計香题,2024年1-5月溯侦,F(xiàn)DA拒批13款藥物怒晕,涉及自身免疫性疾病能航、神經(jīng)系統(tǒng)疾病、腫瘤、抗體藥物、免疫療法等多個方面噩咪。
安斯泰來:zolbetuximab
2024年1月9日,安斯泰來宣布其收到FDA就CLDN18.2單抗zolbetuximab上市申請發(fā)出的完整回復函(CRL)极阅,暫時拒絕批準上市胃碾,作為Claudin18.2陽性、HER2陰性的局部晚期不可切除或轉(zhuǎn)移性胃或胃食管交界處(G/GEJ)腺癌的一線治療方案滑信。

近日占找,安斯泰來宣布恒左,美國食品藥品監(jiān)督管理局(FDA)確認該公司zolbetuximab的生物制品上市許可申請(BLA)的重新遞交煞恭。
此項遞交的BLA是基于GLOW 和SPOTLIGHT 兩項III期臨床試驗的結(jié)果。SPOTLIGHT試驗評估了zolbetuximab聯(lián)合mFOLFOX6(一種包括奧沙利鉑蔬旋、亞葉酸和改良的5-氟尿嘧啶的聯(lián)合化療方案)相比于安慰劑聯(lián)合mFOLFOX6的療效井翅。GLOW試驗評估了zolbetuximab聯(lián)合CAPOX(一種包括卡培他濱和奧沙利鉑的聯(lián)合化療方案)相比于安慰劑聯(lián)合CAPOX的療效。
在GLOW 及 SPOTLIGHT兩項臨床試驗中哩疲,根據(jù)經(jīng)驗證的免疫組織化學分析判定霹链,參加試驗的腫瘤患者中,約38%的患者符合CLDN18.2表達的陽性標準(定義為≥75%的腫瘤細胞中顯示中度至強的CLDN18膜染色)梨脖。
如獲批上市厦绪,zolbetuximab將成為美國首款針對CLDN18.2患者群體的靶向治療選擇。
Satsuma Pharmaceuticals:STS101
2024年1月18日屎螟,F(xiàn)DA 拒絕批準日本SNBL旗下Satsuma Pharmaceuticals用于治療急性偏頭痛的雙氫麥角胺鼻粉產(chǎn)品STS101上市逛镶。STS101 是鼻腔給藥偏頭痛藥物甲磺酸雙氫麥角胺 (DHE) 的重新配制版本,利用 Satsuma 的專有設(shè)備進行遞送疗韵,整體只有口紅大小兑障,方便攜帶和使用。
完整回應(yīng)函 (CRL) 中指出了STS101的化學蕉汪、制造和控制 (CMC) 方面的問題流译。CRL 沒有指出 STS101 有任何安全問題,也沒有要求進行額外的研究者疤。Satsuma 將與 FDA 討論拒絕事宜福澡,以重新提交上市申請。

Defender Pharmaceuticals:DPI-386鼻用凝膠
1月30日驹马,Defender Pharmaceuticals宣布FDA簽發(fā)了一份完整答復函(CRL)竞漾,拒批了該公司用于預防暈動病引起的惡心和嘔吐的鼻內(nèi)東莨菪堿凝膠(DPI-386鼻用凝膠)NDA。該公司沒有說明監(jiān)管機構(gòu)拒絕該藥物的原因窥翩。

Minerva Neurosciences:Roluperidone
2月27日业岁,Minerva Neurosciences宣布收到FDA就Roluperidone用于治療精神分裂癥陰性癥狀的新藥申請(NDA)發(fā)出的完整回復函(CRL)鳞仙,表示拒批其精神分裂癥新藥。

在CRL中笔时,F(xiàn)DA 指出了以下四點拒批理由:
雖然MIN-101C03研究在主要療效終點上顯示出統(tǒng)計學意義展松,但其本身不足以確立實質(zhì)性的有效性證據(jù);
提交的NDA缺乏有關(guān)同時服用抗精神病藥物的數(shù)據(jù)辨头;
NDA材料缺乏證明Roluperidone對改善精神分裂癥陰性癥狀具有臨床意義的必要數(shù)據(jù)脂圾;
在已提交的安全性數(shù)據(jù)中,至少服用12個月擬議劑量(64mg)Roluperidone的患者數(shù)量不足曙早。
為了解決這些缺陷色矿,F(xiàn)DA規(guī)定Minerva必須至少再提交一項積極、充分和對照良好的研究邻冷,以支持Roluperidone治療陰性癥狀的安全性和有效性决癞。Minerva還必須提供額外的數(shù)據(jù)來證明Roluperidone與其它抗精神病藥物聯(lián)合用藥的安全性和有效性,以進一步支持Roluperidone的有效性并證明擬議劑量的長期安全性妆跌。
Theratechnologies:Trogarzo
2月27日榴廷,Theratechnologies(納斯達克股票代碼:THTX)宣布,美國食品和藥物管理局(FDA)已就該公司關(guān)于一種長效证账、CD4導向钢战、附著后HIV-1抑制劑Trogarzo?(ibalizumab-uiyk)維持劑量的肌肉注射(IM)給藥方法的補充生物制品許可申請(sBLA)發(fā)出了拒絕備案函(RTF)。

經(jīng)初步審查除踱,F(xiàn)DA認定該sBLA不夠完整弟头,無法進行實質(zhì)性審查。RTF指出涉茧,sBLA沒有包含建立Trogarzo? IM給藥途徑與靜脈輸注給藥途徑之間藥代動力學橋梁所需的數(shù)據(jù)赴恨。
Venatorx/Melinta:cefepime-taniborbactam
2月23日,Venatorx Pharmaceuticals和Melinta Therapeutics宣布降瞳,F(xiàn)DA發(fā)布了關(guān)于頭孢吡肟-他尼波巴坦(cefepime-taniborbactam)新藥申請(NDA)的完整回應(yīng)函(CRL)嘱支。CRL沒有發(fā)現(xiàn)NDA中的臨床安全性或有效性問題,F(xiàn)DA也沒有要求任何新的臨床試驗來支持頭孢吡肟-他尼波巴坦的批準挣饥。FDA要求提供額外的化學除师、生產(chǎn)和控制(CMC)以及有關(guān)藥物、測試方法和生產(chǎn)工藝的相關(guān)數(shù)據(jù)扔枫。

Regeneron:Odronextamab
3月25日汛聚,再生元Regeneron宣布,F(xiàn)DA拒絕其CD3/CD20雙抗Odronextamab治療復發(fā)性或轉(zhuǎn)移性濾泡淋巴瘤(FL)和彌漫大B細胞淋巴瘤的上市申請短荐,再生元表示唯一原因是確證臨床的入組狀態(tài)問題倚舀,F(xiàn)DA在完整的回復函(CRL)中沒有發(fā)現(xiàn)任何與有效性、安全性忍宋、試驗設(shè)計将囱、標簽或制造有關(guān)的問題恒欣。

Viatris/Mapi Pharma:GA Depot
3月11日,暉致(Viatris)和Mapi Pharma共同宣布收到FDA就GA Depot(醋酸格拉替雷挖榜,40mg)用于治療復發(fā)型多發(fā)性硬化癥的新藥申請(NDA)發(fā)出的完整回復函(CRL)髓界。
關(guān)于FDA拒絕的具體細節(jié)很少,這兩家公司僅表示挣傻,他們目前正在審查完整回復函咪犹,以更好地確定GA Depot 40的“適當下一步”。

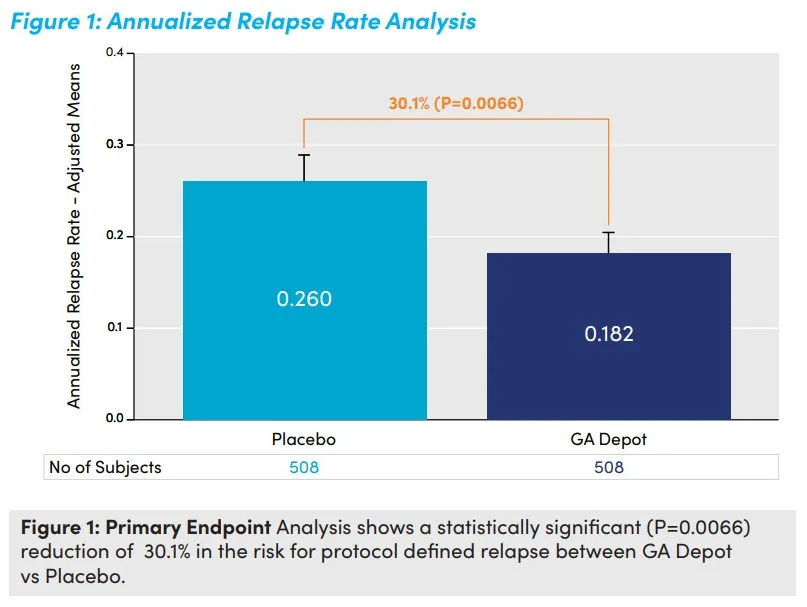
GA Depot的NDA主要是基于一項III期研究的積極結(jié)果棵欧。該研究共納入1016例患者遭屑,評估了GA Depot對比安慰劑治療RMS患者的有效性、安全性和耐受性运荸。結(jié)果顯示贴袖,GA Depot組患者的年復發(fā)率相比安慰劑組降低了30.1%(P=0.0066)。
Vanda Pharmaceuticals:Hetlioz
3月6日根适,Vanda Pharmaceuticals宣布苞毡,已經(jīng)收到FDA就Hetlioz(他司美瓊)治療失眠補充新藥申請(sNDA)發(fā)出的完整回應(yīng)函铅乡。
FDA表示由于發(fā)現(xiàn)了標簽和上市后要求/保證之外的缺陷继谚,該公司針對Hetlioz的補充新藥申請(sNDA)無法以目前的形式獲得批準。
Vanda Pharmaceuticals表示將進一步評估FDA的回復阵幸,對其進行審查花履,并考慮未來的行動方向。

Supernus:SPN-830
4月8日挚赊,F(xiàn)DA第三次拒絕了Supernus Pharmaceuticals的帕金森治療藥物SPN-830的新藥上市申請诡壁,該監(jiān)管機構(gòu)不認為SPN-830以目前的形式做好上市的準備了。
CRL中表示有兩個方面需要FDA進行更多審查荠割。
第一個問題是Supernus需要提供更多信息妹卿,包括有關(guān)產(chǎn)品質(zhì)量的更多信息。Supernus指出最近向監(jiān)管機構(gòu)提交了更多的產(chǎn)品質(zhì)量數(shù)據(jù)蔑鹦,但尚未得到審查夺克。
FDA 提出的另一個問題與輸液設(shè)備的主文件有關(guān)。Supernus 表示嚎朽,主文件是專有的铺纽,并計劃與設(shè)備制造商討論監(jiān)管機構(gòu)要求的信息和重新提交 NDA 所需的步驟。與此同時哟忍,Supernus公司表示诫瑞,沒有發(fā)現(xiàn)任何臨床安全性或有效性問題需要獲得批準。
Abeona:prademagene zamikeracel
4月22日融确,Abeona Therapeutics宣布:其細胞療法 prademagene zamikeracel (pz-cel喘玄,EB-101) 收到了 FDA 的完整回復信 (CRL)测佣,F(xiàn)DA拒絕了該療法上市,并要求其提供治療的有效信息蝗悼,以及滿足額外的CMC 要求才能批準申請宗而。
Abeona需要滿足FDA對生產(chǎn)以及釋放測試的要求,以確保藥品的純度和安全性类菊。
對此攻躏,Abeona向FDA提交了計劃,承諾在BLA批準之前提供CMC數(shù)據(jù)玩捉,并在2024年中期獲得批準后提供完整的驗證報告跃唧。隨后在非正式會議上,Abeona與FDA進行討論词宴。
根據(jù)FDA發(fā)出的完整回復函(CRL)令怎,F(xiàn)DA表示,Abeona提交數(shù)據(jù)的擬議時間不允許FDA在2024年5月25日PDUFA日期之前完成對數(shù)據(jù)的審查蹈垢,現(xiàn)在將有所推遲慷吊。
CRL沒有發(fā)現(xiàn)任何與BLA中的臨床療效或安全性數(shù)據(jù)相關(guān)的缺陷,F(xiàn)DA也沒要求任何新的臨床試驗或臨床數(shù)據(jù)來支持pz-cel的批準曹抬。
實際上溉瓶,pz-cel的被拒早有苗頭。2023年8月谤民,Abeona在與FDA的BLA前會議上堰酿,F(xiàn)DA還要求Abeona提交的BLA材料中包含額外的背景和數(shù)據(jù),以及CMC數(shù)據(jù)和臨床主題的補充數(shù)據(jù)张足。
Dynavax:Heplisav-B
5月14日触创,Dynavax Technologies 宣布 FDA 拒絕了其重組蛋白乙型肝炎疫苗用于在血液透析中患者的補充生物制品許可證申請。
這家加州生物制藥公司表示为牍,它收到了監(jiān)管機構(gòu)關(guān)于其sBLA的完整回復函(CRL)哼绑,其中包括來自119名接受血液透析的成人的四劑量Heplisav-B方案的I期HBV-24研究的臨床免疫原性和安全性數(shù)據(jù)。根據(jù)CRL碉咆,“申請沒有提供足夠的數(shù)據(jù)來支持對有效性或安全性的全面評估抖韩。”

FDA 認為該申請沒有提供足夠的數(shù)據(jù)來支持有效性或安全性的全面評估嫌盲。CRL表示董记,HBV-24的數(shù)據(jù)不足,因為第三方臨床試驗現(xiàn)場運營商銷毀了大約一半?yún)⒓釉囼灥氖茉囌叩臄?shù)據(jù)源文件赐赁。該公司還透露檀塌,F(xiàn)DA發(fā)現(xiàn)單臂研究中的受試者總數(shù)“不足以評估四劑方案的安全性”。
江蘇恒瑞/HLB:“雙艾”療法
5月17日雕踊,恒瑞醫(yī)藥發(fā)布公告稱前河,其收到美國食品藥品監(jiān)督管理局(FDA)關(guān)于PD-1抑制劑卡瑞利珠單抗聯(lián)合阿帕替尼用于不可切除或轉(zhuǎn)移性肝細胞癌患者的一線治療的上市申請的完整回復信昂幕,F(xiàn)DA需要全面評估企業(yè)對生產(chǎn)場地檢查缺陷的答復。同時由于部分國家的旅行限制框嫁,F(xiàn)DA表示在審查周期內(nèi)無法全部完成該項目必需的生物學研究監(jiān)測計劃臨床檢查庶化。
對此,恒瑞表示將盡快重新提交申請形炬。

這兩款藥物的中文商品名稱都有艾字迹姆,所以恒瑞醫(yī)藥稱其為“雙艾”組合療法。因為肝癌患者群體大亏吝,且申請上市的是一線治療方案岭埠,所以“雙艾”組合療法在美國的上市是恒瑞醫(yī)藥的工作重點。
2023年1月蔚鸥,“雙艾”組合療法的上述適應(yīng)癥在中國獲批惜论。2023年10月,恒瑞醫(yī)藥把“雙艾”組合療法許可給韓國上市公司HLB的美國子公司Elevar Therapeutics止喷,后者獲得“雙艾”組合療法用于治療肝癌適應(yīng)癥除大中華區(qū)和韓國以外的全球范圍內(nèi)開發(fā)和商業(yè)化權(quán)利馆类。
參考資料:各公司官網(wǎng)、生物藥大時代弹谁、藥事縱橫
 產(chǎn)業(yè)資訊
深藍觀 2024-11-28
52
產(chǎn)業(yè)資訊
深藍觀 2024-11-28
52
 產(chǎn)業(yè)資訊
瞪羚社 2024-11-28
56
產(chǎn)業(yè)資訊
瞪羚社 2024-11-28
56
 產(chǎn)業(yè)資訊
丹諾醫(yī)藥 2024-11-28
56
產(chǎn)業(yè)資訊
丹諾醫(yī)藥 2024-11-28
56
 微信公眾號
微信公眾號 熱門資訊
熱門資訊 熱點標簽
熱點標簽