

 政策法規(guī)
政策法規(guī)
 2013-03-01
2013-03-01
 4352
4352
來源:國家食品藥品監(jiān)管局藥品審評中心 2013-2-28
?
2012年度中國藥品審評報告
國家食品藥品監(jiān)督管理局藥品審評中心
發(fā)布日期:20130228
2012年8月31日德國西部城市施托爾貝格豎起了一尊銅像,名為“生病的孩子”闷墅。銅像左邊是一個沒有雙臂幔托、雙腿畸形的小女孩靠在一張椅子上,右邊是一張空椅子,銅像底座中間寫著“紀念那些死去的和幸存的沙利度胺受害者”穷抹。這一年相梭,是反應停(沙利度胺)事件50周年!
這是一起轟動全球的慘労堆础党饮!上世紀50年代,德國一家制藥商推出鎮(zhèn)靜劑反應停驳庭。這種藥品對減輕婦女懷孕早期出現的惡心刑顺、嘔吐等反應有效,于是迅速在多個國家推廣饲常。而此時蹲堂,美國食品藥品監(jiān)督管理局(FDA)一位叫弗蘭西斯·凱爾西的審評員也在案頭審查這家德國公司的資料。由于凱爾西博士發(fā)現資料中有許多不確定的數據和其它問題,反應停未被準許在美國上市柒竞。后來政供,使用該藥品的歐洲、澳大利亞能犯、加拿大和日本等先后發(fā)現了新生兒先天四肢殘缺鲫骗,即海豹嬰兒。經科學證實踩晶,新生兒四肢殘缺的罪魁禍首就是反應停执泰。至此,全球已有近一萬名左右因反應停引發(fā)的海豹兒出生子桩。
正是這場慘劇推動了歐美各國自上世紀60年代起不斷變革杖烘、完善藥品上市的科學審查制度。
反應停的慘劇發(fā)生時奄刊,我國醫(yī)藥領域與歐美國家?guī)缀鯖]有接觸因而免于此害于抬,但同時我國藥物的研發(fā)基本上是仿制一些藥物,藥品的供給能力有限饱粟,缺醫(yī)少藥問題十分突出元邻。
半個多世紀過去了,世界發(fā)生了巨大的變化掖阶,藥品的研發(fā)和市場供應已實現全球化咒付。歐美等藥品的上市審評制度在經歷了慘劇后,痛定思痛炮疲,逐漸形成了現有的國家立法枷斩、政府立規(guī)、專業(yè)機構審評的制度體系虎叔,并已在審評方法和手段上建立了完備的科學體系味测。我國自建國以后建立了藥品標準管理制度、藥品檢驗制度铺坞,于80年代中期建立了上市藥品的審評制度起宽。隨著我國衛(wèi)生事業(yè)的發(fā)展和醫(yī)藥經濟的進步,這一制度也在不斷發(fā)展和完善康震。
當前燎含,在我國承擔藥品上市審評的機構為:國家食品藥品監(jiān)督管理局藥品審評中心。其主要職責有兩個方面:一是負責藥物(藥品在上市前被稱為藥物)進行人體臨床試驗前的科學性審評腿短,以保證臨床試驗是在受試者有安全保障、人體研究符合倫理和科學標準等條件下進行绘梦;二是負責藥品上市前的科學性審評橘忱,保證公眾用藥的安全、有效和產品質量的可控。藥品審評要對申請的藥物是否具有臨床治療價值钝诚、患者的需求是否得到滿足颖御、公眾能否用得上與用得起、藥物的風險是否被充分認知凝颇、控制風險的手段是否可行潘拱、臨床受試者是否得到充分的保護、藥物的有效性是否確切末瘾、藥物的質量是否具有控制手段等多種復雜的因素唠芋,進行綜合分析評判。這些分析評判必須依賴翔實的科學證據讼舰,需要審評人員清晰的邏輯分析和專業(yè)能力支持拧淘,需要各種無利益沖突的專家的參與,也需要同藥物研發(fā)者平等交流獲取信息和知識沦望。評判必須在患者獲益和風險之間平衡族焰。每一個審評結論,“批準”或者“不批準”慨醒,都是對這個機構能力的一次挑戰(zhàn)蚊凫!
藥品審評中心的使命:維護和促進公眾健康。
藥品審評中心的發(fā)展愿景:值得信賴的阐合、有國際水平的公眾健康的守護者肛英。
藥品審評中心的工作標準:質量、效率颤与、透明水导、清晰、一致和可預見继找。這與各國審評機構普遍認可的《藥品審評質量管理規(guī)范》(GRP)所確立的核心標準是一致的遂跟。
我們清晰地認識到,藥品審評中心目前的能力與所承擔的責任是有距離的婴渡。我們在審評機構內倡導這樣的價值觀:開放幻锁、創(chuàng)新、公正边臼、實證哄尔、團隊、責任柠并。這也是機構的使命和機構的發(fā)展歷程所凝聚的組織文化要素岭接。基于現實臼予,我們提出以受監(jiān)督鸣戴、可評估啃沪、能發(fā)展為推進機構發(fā)展的基本要求,借鑒國際法規(guī)科學的發(fā)展經驗窄锅,著力提升科學審評的能力创千,加強專業(yè)化審評制度的建設,切實保障公眾用藥安全割对、維護和促進公眾健康揖姆。
我們愿以開放的態(tài)度接受社會各界的監(jiān)督,現將2012年度中國藥品審評報告呈現給公眾援漓。
一眼厕、2012年重要工作舉措
2012年,藥品審評中心在諸多方面采取措施彼使,探索前行织鳖,更好地服務于公眾健康需求。
(一)按照藥物研發(fā)規(guī)律接馏,調整審評策略
鼓勵創(chuàng)新研發(fā)以解決未被滿足的臨床需求卷哟。促進臨床短缺的仿制藥研發(fā)以解決公眾用藥的可及性和可支付性問題,是全球藥品審評機構普遍關注的問題扇蚯。創(chuàng)新藥和仿制藥的研發(fā)有其各自不同的特點役躬,審評機構應根據它們各自的研發(fā)規(guī)律,采取不同的審評策略柿糖,保證審評的科學性和效率纸兔。
關于創(chuàng)新藥的審評
當今全球藥物創(chuàng)新的首要領域就是解決未被滿足的臨床需求。藥物創(chuàng)新進程中必須經歷“臨床準入”和“市場準入”這兩個環(huán)節(jié)否副。審評機構在這兩個環(huán)節(jié)中發(fā)揮著鼓勵創(chuàng)新和控制風險并重的作用汉矿。
2012年,藥品審評中心為發(fā)揮好上述兩方面的作用备禀,采取了以下主要措施:
——鼓勵以臨床價值為導向的藥物創(chuàng)新洲拇,發(fā)揮好審評的導向作用。鼓勵創(chuàng)制針對我國重大疾病譜具有較好治療作用的藥物曲尸。鼓勵兒童用藥赋续、罕見病藥物的研發(fā)。上述范圍的創(chuàng)新藥審評任務被列為優(yōu)先級任務另患,實行全程督導管理纽乱。同時,與重大專項實施管理辦公室及時溝通昆箕,加快國家重大專項品種的審評鸦列,使審評工作更好地落實國家藥物創(chuàng)新戰(zhàn)略。
——啟動《申請概述》撰寫工作鹏倘。圍繞臨床方案開展“靶向審評”升筛,以變革研發(fā)和審評從仿制藥沿襲的慣性思維振沾。針對創(chuàng)新藥首次臨床試驗申請垛搏,臨床專業(yè)審評人員一般會在審評中心承辦任務后1個月內柳竟,在圍繞臨床試驗方案開展全面評價之前,先行對申請內容肩卡、所申請適應癥的現有治療手段進行概括性評價慰乾,重點關注申請的臨床價值,以及臨床試驗方案的基本情況嗦忍,以幫助確定審評任務的優(yōu)先級殊童,更重要的是幫助安全性評價、藥學評價確定合理的技術要求阿绣,例如長期毒性研究海泵、穩(wěn)定性研究的要求等。
——建立基于創(chuàng)新藥物開發(fā)階段的藥學審評及動態(tài)跟進的審評模式郁邪,徹底轉變了創(chuàng)新藥藥學審評和研發(fā)的思路围婴。研發(fā)各個階段的特點,建立創(chuàng)新藥臨床前藥學評價模板和研發(fā)期間的年度報告制度昧识,藥學方面的技術要求與國際基本接軌钠四,并使藥學更新或變更的資料能夠滾動提交。上述措施在2012年5月正式實施后跪楞, 創(chuàng)新藥的藥學審評不再成為審評的瓶頸缀去。
——基于風險可度量、可評估甸祭、可預測缕碎,探索建立創(chuàng)新藥臨床準入和上市準入的風險管理模式。以實施有效的風險管理為目標池户,根據創(chuàng)新藥所處研發(fā)階段以及上市后臨床使用條件下的獲益風險評估咏雌,分別以臨床批件、審評建議函煞檩、生產上市批件(進口批件)处嫌、風險控制計劃、說明書等斟湃,對申請人熏迹、臨床研究者、合同研究組織(CRO)等提出相應的風險管控要求凝赛。特別是針對創(chuàng)新藥臨床試驗挫勿,讓參與人體研究的各相關方共同管控風險,切實保護臨床受試者的權益庭授。
——發(fā)布《臨床試驗數據管理工作技術指南》琴哗,開通藥物臨床試驗登記和信息公示平臺绎探,以保證臨床研發(fā)符合國際公認的數據管理準則。按照國際標準構建了我國藥物臨床試驗的數據管理和數據標準化工作平臺吟叙,為進一步加強臨床試驗全過程闲堆、動態(tài)監(jiān)督管理、保證試驗數據的真實揍丘、規(guī)范捍辫、可溯源奠定了基礎。
——變革溝通交流的模式楞庸,制訂實施《藥品審評中心與申請人溝通交流質量管理規(guī)范》沉年,以保證中心的決策是在全面信息采集下進行,提升決策的質量和效率晾胡。申請人與中心均可就研發(fā)中的關鍵技術問題提出溝通交流脾婚,交換信息和觀點,并形成雙方簽字認可的會議紀要罢猪。2012年近她,藥品審評中心與申請人共召開了37 次溝通交流會,形成了37份會議紀要坡脐。增加與申請人交流的窗口泄私,開通申報資料滾動提交通道,減少既往只能以“發(fā)補”方式完善資料所帶來的不必要的排隊時間备闲。創(chuàng)新藥臨床試驗申請的發(fā)補率由2011年的41.1%降低至2012年的33.9%晌端。
——鼓勵體現中藥臨床特點的創(chuàng)新。按照中藥的研發(fā)的規(guī)律和特點恬砂,進一步探索建立現代中藥有效性及安全性的評價體系咧纠。組織召開了中藥新藥研究與評價研討會,邀請了國內知名專家共同探索中藥創(chuàng)新研究的方向和思路泻骤。一是在中藥新藥的審評中漆羔,充分考慮其臨床應用價值及療效特點,加強對于中藥新藥臨床有效性假設及研究邏輯的審評狱掂,推動中藥臨床試驗從驗證性研究向探索性研究的轉變演痒;二是針對中藥成分復雜,需要全過程控制質量的特點陈伪,強調以保證藥品質量的穩(wěn)定均一為核心深牲,要求工藝研究應充分反映規(guī)模化生產的可行性泛倦;三是工藝研究應與臨床療效和質控體系結合辽松,保證上市后產品批間質量的一致性。
——中藥實施分類審評习环。把握不同類型中藥的特點裸努。對于有效部位芦终、有效成分新藥,重點關注其立題依據及安全性研究垃桨;對用于中醫(yī)優(yōu)勢病種及非優(yōu)勢病種的中藥復方新藥眉堪,根據其人用經驗,探索進行有效性的差異化評價版逼;對于中藥注射劑赌列,嚴格控制安全風險,體現臨床優(yōu)勢钮隙。
關于仿制藥的審評
當前,無論是發(fā)達經濟體還是新興經濟體國家都十分重視仿制藥的研發(fā)属瓣,以最大限度地解決國家醫(yī)藥支付體系和公民個人在藥品可及性和可支付性上的負擔载迄。審評機構在仿制藥的市場準入中發(fā)揮著重要的評判作用,評判其是否與原研產品具有一致性和可替代性抡蛙。
由于各種原因护昧,我國仿制藥申請量高、仿制藥重復申請嚴重粗截、工業(yè)化能力不足等問題尚未得到根本解決惋耙。同時又存在部分臨床治療價值高、臨床亟需仿制藥供應不足的問題熊昌。為更好滿足臨床需求绽榛,促進我國仿制藥的健康發(fā)展,藥品審評中心在仿制藥領域探索了一系列的改革措施:
——對我國仿制藥的研發(fā)和審評現狀加以診斷婿屹。研究發(fā)布《中國通用名藥發(fā)展研究報告——市場準入制度研究(2012年)》灭美。由國家藥品審評中心和軍事醫(yī)學科學院毒物藥物研究所發(fā)起、國家科技部批準成立的“通用名藥物品種產業(yè)技術創(chuàng)新戰(zhàn)略聯盟”發(fā)布了中國通用名藥2012年的研究報告(研究報告全文下載地址: http://www.cde.org.cn/news.do?method=viewInfoCommon&id=312869)昂利。該研究報告基于對我國仿制藥的發(fā)展現狀和發(fā)展路徑的分析研究届腐,重點診斷分析了仿制藥“市場準入制度”存在的關鍵問題,系統(tǒng)提出了以“滿足公眾健康需求榕哩、滿足國家戰(zhàn)略需求”為核心的關于我國仿制藥準入制度的完善建議仪荞。
——探索仿制藥優(yōu)先審評的機制,推進有臨床治療價值督近、臨床亟需仿制藥可及性問題的解決江构。基于試點實踐屿赶,提出了納入優(yōu)先審評仿制藥的基本條件覆珍,即一是具有較高臨床價值,且臨床需求尚未有效解決差机;二是實現工業(yè)化生產纱轨、能夠形成有效供給耿愈;三是按照國際技術標準建立全面、系統(tǒng)的藥品質量控制體系倔剩。在國家局的領導下朦暖,與多部委溝通,形成了優(yōu)先審評仿制藥目錄的產生機制盖呼,構建了優(yōu)先審評仿制藥的注冊流程儒鹿。
——探索生產現場檢查和技術審評相結合的工作機制,解決仿制藥工業(yè)化能力不足的問題几晤。以替加環(huán)素约炎、地西他濱和卡培他濱等品種為試點,把生產現場檢查融入到審評過程中蟹瘾,派遣一線審評人員參加生產現場檢查圾浅,實現“資料鏈接現場”的審評,增強審評人員對產品和生產工藝的理解憾朴,確保批準的產品具有高質量且可工業(yè)化生產狸捕。
——繼續(xù)推動國際通用技術文檔(簡稱CTD)格式申報,使仿制藥的研發(fā)和審評與國際的技術標準對接众雷。CTD格式不僅是一種模塊化申報資料要求灸拍,他更體現了研發(fā)者的研發(fā)理念、研發(fā)邏輯以及研發(fā)全過程管理水平砾省,同時避免審評者對審評數據的采集丟失鸡岗。藥品審評中心鼓勵申請人按照CTD格式所體現的研發(fā)理念與技術要求開展研究,對采取CTD格式申報的仿制藥單獨排隊纯蛾、優(yōu)先審評肌辑。總結在CTD格式申報資料審評中發(fā)現的共性問題留旱,并在中心網站發(fā)布( http://www.cde.org.cn/dzkw.do?method=largePage&id=312898 )刹造。
2012年,藥品審評中心根據國家有關要求格你,繼續(xù)優(yōu)先審評艾滋病治療藥物劫欣、抗耐藥結核藥物、氟利昂替代等類型的仿制藥述逾。同時盹清,將有限的審評資源向有較高臨床治療價值,且臨床亟需的仿制藥傾斜殴客。
(二)信息公開曾辙,提供公共服務,接受社會監(jiān)督
藥品審評的終端服務對象是患者和公眾,專業(yè)服務對象是臨床醫(yī)生和醫(yī)療機構糕米,直接服務對象是制藥企業(yè)和藥品研究機構忠伊。各方有權利獲知審評的過程和結果。公眾有權利了解自己服用的藥品因何有效巷波,有哪些不良反應萎津,服用時有哪些注意事項;臨床醫(yī)生有權利精準抹镊、全面锉屈、深入的了解藥品的特點、作用機理垮耳、適應癥颈渊、不良反應等,以合理用藥氨菇;研究者儡炼、投資者有權利了解審評的決策依據以及可預期的結果。
審評機構應該把審評決策的科學數據和模型轉化為公眾可以接受的科普語言對全社會公示查蓉,以接受專業(yè)人士、行業(yè)和公眾的監(jiān)督榜贴。2012年豌研,藥品審評中心進一步加強信息公開力度,以更好地服務于社會并接受社會沈猜。
向社會公開方面采取了以下措施:
——在中心網站發(fā)布我國新上市藥品的《審評概述》淌璧。通過《審評概述》,清晰地告知公眾審評機構是基于什么數據和實證做出決策奈炕,批準這個藥物的禽忧。其中包括:這是一個什么樣的疾病,患者承受哪些痛苦书尚;這個疾病的致病因素有哪些衙乡,在全球有無明顯的差異;這個疾病現在有無有效的治療手段沐刷,如有箱歪,現有手段還有哪些不足;這個藥物做了哪些研究验柴,研究的結果證明了在這個疾病的哪些環(huán)節(jié)可以發(fā)揮治療作用揽膏、對患者有明確的益處;這個藥物的研究中暴露了哪些安全性信號姊宗,基于這些安全性歹朵、有效性信息的風險獲益分析,針對這些安全性信號已經采取了哪些控制措施撼泛;還可能有哪些潛在的安全性問題挠说,我們有無相應的手段預警和管控澡谭;這個產品上市后還有哪些其他的要求等。2012年已發(fā)布38篇審評概述纺涤。
——加強藥物臨床試驗的信息公開译暂。在中心網站建立“臨床試驗登記與公示”系統(tǒng)( http://www.cde.org.cn/news.do?method=changePage&pageName=serviceLcsy&frameStr=125)。向社會公示撩炊,哪些藥品已經進入臨床開展人體研究外永;公示每一個研究項目、研究者和地點等信息拧咳,使這項研究活動在社會倫理道德認可的條件下進行伯顶,促進對受試者的保護。
——主動向公共媒體開放骆膝,接受媒體的監(jiān)督祭衩。2012年《科技日報》、《中國醫(yī)藥報》阅签、《醫(yī)藥經濟報》等專業(yè)媒體以及社會科學研究機構參加了中心的開放日掐暮、論壇、專家咨詢會列序、研討班等活動碧爬,從第三方的視野審視藥品審評工作。
向申請人公開方面采取了以下措施:
——公開待審品種排隊序列睹肝。依托中心的網站旺胳,公開了中心承辦的全部注冊申請以及各類化學藥申請的任務序列;讓大家都知道自己申報的品種排在什么位置画柜,預計多長時間能完成審評捞蹈,便于申請人安排工作計劃。
——公開“加快審評”的品種和加快原因围杉。凡屬加快審評的品種钧鸳,都會在中心網站上說明原因,讓公眾了解審評資源傾斜的原因弄讥。
——各月化藥的審評計劃及其完成情況全部上網公開挣呛,定期公示中心審評任務完成情況及審評結論,讓申請人對審評進程進行監(jiān)督檢查谋啃。
——通過“申請人之窗”向申請人公開不批準品種的審評報告舰绘。對于技術審評結論為“不批準”的品種,向申請人全文公布技術審評報告葱椭,申請人全面了解不批準理由捂寿,并可在15日內提出申訴意見。2012年我們已接受了214個不批準品種的申訴意見,并充分考慮申請人的意見后做出審評結論秦陋。
——公布審評咨詢會議信息蔓彩。所有咨詢審評專家的會議情況均在會后予以公示,全年共召開專家咨詢會議11次驳概,涉及189個品種赤嚼,邀請專家1590人次。
——公布復審品種審評計劃和復審結論顺又。2012年全年收審復審申請170個更卒,完成復審任務196個。其中維持原結論161個稚照,占82.1%蹂空;,糾正原審評結論35個果录,占17.9%羹授。
——發(fā)布已有批準文號與在審品種信息。針對仿制藥重復研究咖雀、重復申報的問題擎势,為合理引導藥物研發(fā)的方向,藥品審評中心對已批準上市品種和在審的重復品種進行了全面亮购、系統(tǒng)的整理分析索官,并在藥品審評中心網站發(fā)布( http://www.cde.org.cn/drugInfo.do?method=init)。
——起草滨鼠、修訂藥物研究技術指導原則,發(fā)布技術標準乐标。為指導研發(fā)工作捶臂,清晰闡述藥品研發(fā)的技術標準和要求,2012年姚损,藥品審評中心繼續(xù)完善藥品研發(fā)的技術指導原則體系赎躲,啟動起草36項、修訂7項指導原則紊徊,已有3項由國家食品藥品監(jiān)督管理局正式發(fā)布實施肴熏,8項公開征求意見。此外顷窒,針對研發(fā)中存在的一些具體技術問題蛙吏,在中心網站開辟了“共性問題解答”專欄,解答103條問題鞋吉;針對研發(fā)和審評中新問題的研究鸦做,在中心網站電子刊物專欄發(fā)表了40篇文章,提出對這些問題的前瞻性思考。
——舉辦第三屆中國藥物創(chuàng)新論壇泼诱√陈樱基于對國內外近800家企業(yè)的調研數據,分析國內研發(fā)模式治筒、研發(fā)能力和風險控制能力屉栓,探討我國藥物審評應對策略。
——繼續(xù)采用多種方式開展溝通交流耸袜。全年舉辦8次開放日活動友多,向公眾介紹中心的基本情況,參加人員達236人次辣摘。舉辦16期藥品技術評價研討班撼由,與業(yè)界共同探討藥品研發(fā)和評價的技術要求與標準,共有6829人次的研發(fā)人員參與涵群。另外狭鳖,通過中心網站的信息反饋平臺和主任信箱,解答533 個問題盲拐。
(三)引入評估機制拳沙,著力提高自身能力
——針對“中國藥品審評制度定位與作用發(fā)揮研究”,中心委托清華大學公共管理學院和哈佛大學醫(yī)學院中國項目部号均,成立聯合課題組怔惯,對我國藥品審評體系的目標定位、藥品審評的法規(guī)體系易震、藥品審評制度與機制以及審評機構的專業(yè)能力建設等問題庞取,進行診斷和分析,2012年已經完成了專題研究烈瑰。
——針對審評機構決策質量和審評質量管理體系建設問題鞍靴,中心邀請國際法規(guī)科學創(chuàng)新研究中心(CIRS),對中心質量管理體系進行了診斷和分析镶摘。2012年5月嗽桩,CIRS與中心就審評質量管理體系(Good Review Practice ,GRP)的實施進行了交流凄敢,并進行基線調查碌冶。9月又對中心管理團隊進行了問卷調查和社會調查。11月向中心報告了初步評估意見涝缝。本次評估是CIRS首次在亞太地區(qū)開展的機構內的正式評估扑庞。
——針對疫苗審評質量管理體系建設,并結合WHO對疫苗監(jiān)管體系評估工作的要求俊卤,推進ISO-9001評估和認證工作嫩挤。2012年8月開展了內審員培訓害幅,9月完成對《質量手冊》和《程序文件》的起草和修訂工作,并請各部門制定好相關三級文件岂昭。目前以现,中心已進入疫苗質量管理體系運行階段,并進入ISO-9001認證二期審查约啊。
——針對審評機構的審評能力和工作效率邑遏,中心制定出臺了《審評人員職務調整考核評估管理辦法》和《績效考核工作辦法》,以定量的指標和數據來評估中心的工作效率瘫笋。
(四)加強制度建設路旬,推動事業(yè)發(fā)展
藥品審評機構的角色是公眾健康的守護者和促進者。我們要不斷提升機構能力锨堵,以滿足十三億公眾的健康需求哑回;我們是創(chuàng)新的協(xié)同推動者,既要協(xié)同推進創(chuàng)新粟墩,又要有效地控制創(chuàng)新中的風險伊哮,同時還不能成為創(chuàng)新中的障礙。這對我們有相當大的挑戰(zhàn)惨槐。
穩(wěn)健的發(fā)展问嬉,需要制度保障。中心基于現實瘤褒,制定和完善了管理制度和審評流程炮姑,構建專業(yè)化審評制度體系框架。在《藥品審評原則和程序》總原則下悲组,制訂出臺了《藥品審評中心技術審評決策路徑管理規(guī)范》检痰、《藥品審評中心審評任務管理規(guī)范》、《藥品審評中心審評卷宗管理規(guī)范》锨推、《藥品審評中心與注冊申請人溝通交流質量管理規(guī)范》等一系列共10個規(guī)范攀细,不但注重程序審評,更加注重基于證據的審評爱态。以質量、效率境钟、透明锦担、清晰、一致和可預見作為審評工作原則慨削,并將其作為評價審評決策質量的指標洞渔。
穩(wěn)健的發(fā)展,需要一支“高水平缚态、國際化磁椒、有權威”的人才隊伍堤瘤。任何決策都是依賴于人的,只有高素質的人浆熔,才可能有高質量的決策本辐。對于我們這樣一個人力資源有限,卻承擔著13億人口上市藥品把關責任的機構矢匾,對于高素質人才的需求就更加迫切懂崭。我們采取了以下措施:
——探索新的人才引入機制,加大內部人才培訓区戚、培養(yǎng)和考核力度处淘。2012年首次面向社會公開招聘了3名高級審評員;開展專業(yè)挣堪、管理栈眉、英語等方面的培訓工作,培訓人員60余人次豁惨。
——加強技能培訓榛架。加強溝通交流的技能培訓,分內部和外部兩個方面翰鬓。內部的培訓重點在于解決團隊中忽畏、專業(yè)內部、跨專業(yè)和跨組織部門之間的交流存在的障礙坟翠,以保證高質量赖钞、高效率地決策。針對外部交流的培訓重點聘裁,是在于學會傾聽雪营、分析、溝通衡便。準備好傾聽公眾献起、行業(yè)、醫(yī)師镣陕、媒體的意見谴餐,分析他們的訴求,傳播評價機構的理念呆抑。
——加強國際交流與合作岂嗓,借鑒國際先進經驗。在第四屆DIA中國年會舉辦藥品審評中心專場討論會鹊碍,中心整體介紹了我國藥品審評的技術標準厌殉、當前審評和研發(fā)所關注的熱點技術問題和未來發(fā)展趨勢等;與美國FDA同仁會舉辦了“第四屆藥物審評科學決策學術研討會”侈咕,通過模擬的“多學科聯席會”选浅,開展科學論證及討論屏酌,明確建立內部咨詢機制的重要意義,提高審評團隊發(fā)現問題尚技、聚焦問題和解決問題的能力予问。
穩(wěn)健的發(fā)展,需要吸納社會優(yōu)質資源嘲孙,保持審評工作的開放性啤糙。
——繼續(xù)實施并完善專家咨詢制度。在咨詢會議中魂姆,重點關注專家的公正和會議質量問題锉累,解決好利益沖突和保密的問題;在疑難問題上澜茁,充分聽取專家的意見猩蓝;重大品種的決策嘗試采用投票的方式進行集體決策。
——與中華醫(yī)學會相關專業(yè)委員會建立工作機制从肮,加強對臨床急需產品信息的獲取筐子。初步嘗試與呼吸專業(yè)委員會、泌尿專業(yè)委員會 缓苛、血液病專業(yè)委員會等建立聯系芳撒,請專業(yè)委員會確認,在哪些疾病的治療上未桥,還缺少有效的笔刹、老百姓能買得起的藥品,我們將這些藥品加快審評冬耿,盡快滿足公眾的用藥需求舌菜。
——提高審評的開放性,在嚴格資質篩查亦镶、利益沖突規(guī)避制度日月、品種篩選管理和保密制度的保障下,廣泛吸納國內外專家資源缤骨,參與審評的討論與決策爱咬。如在非臨床安全性評價方面,試點合同式購買服務绊起;聘請生產一線藥學專家作為“客座審評專家”參與仿制藥審評台颠。
二、2012年批準重要治療領域藥品情況
2012年搬雳,經過藥品審評中心的審評,提出建議批準以下多個重要治療領域的藥品锤观,為患者獲得最新治療手段提供了可能性晃键,為患者用藥可及性及可支付性提供了重要保證特棕。
(一)抗艾滋病藥物(HIV)領域
針對HIV耐藥以及增加治療的順應性方面,今年提供了以下與全球同步的迟赶、最新的治療手段创哩。
1.利匹韋林片(批準文號:H20120561),是一種二苯胺嘧啶衍生物堵但,屬于新型的非核苷類逆轉錄酶抑制劑(NNRTI)波笆,具有很強的抗野生型HIV-1和耐藥突變株活性。對耐藥的HIV患者具有治療作用铁厌。FDA批準時間2011年5月褂苔。2012年我國批準該藥品上市,使中國患者及時獲得這一最新治療手段惹模。
2.恩曲替諾福韋吡呋酯片(批準文號:H20120568)贩贵,為恩曲他濱和富馬酸替諾福韋二吡呋酯組成的復方片劑,是國際HIV-1感染治療指南推薦的新的治療策略灵妨。復方制劑的引入解阅,增加HIV患者治療的順應性,這對加強治療效果具有重要意義泌霍,為我國控制HIV提供了新的重要手段货抄。
(二)罕見病治療領域
1.注射用地西他濱(批準文號:H20123294,H20120066朱转, H20120067)蟹地,是治療骨髓增生異常綜合癥(MDS)藥物,該病癥是一種罕見病肋拔,既往我國僅有進口產品锈津。2012年,通過建立“資料鏈接現場”機制凉蜂,加強對申請人工業(yè)化生產能力和質量管控能力的考察琼梆,并結合第三方驗證證實產品具有較高的質量水平,先后批準了國內兩個公司的產品生產上市窿吩。
2.蘋果酸舒尼替尼膠囊(批準文號:H20100776-783)茎杂,通過抑制多個靶點而產生抗腫瘤作用和抗血管生成作用藥物。2012年批準該藥品新適應癥瘫篮,用于胰腺神經內分泌腫瘤患者的治療扯连,該病的發(fā)病率低(約為0.3/10萬),發(fā)病后患者極為痛苦唆皇,且不可手術切除梨浑,轉移率高,目前無有效的治療手段吨肆。該藥是在全面考察全球臨床研究數據的基礎上迂儡,采取豁免注冊臨床試驗的審評策略批準上市的芙捏。
(三)兒童用藥領域
1.枸櫞酸咖啡因注射液(批準文號:H20130109),為國際上唯一被批準的治療早產兒呼吸暫停的藥物杰打。早產兒呼吸暫停是一種可能致殘和致命的疾病粤唤,我國既往的醫(yī)療實踐缺乏有效的治療藥物。在全面考察全球臨床數據后搁鞭,采用豁免注冊臨床的審評策略预锅,為此類患兒的生命搶救及時提供了新的治療手段。
2.九味熄風顆粒(批準文號:Z20120034),為首個用于小兒抽動癥的中藥復方制劑恢恼。該適應癥為兒童常見病民傻,近年來發(fā)病率逐漸提高。目前厅瞎,這一治療領域的主要治療藥物均為化學藥物饰潜,如氟哌定醇等,化藥治療的不良反應較多和簸,有些反應患兒多難于接受彭雾。對于輕中度患兒,該中藥制劑顯示出一定的療效锁保,且不良反應較少薯酝,容易被患兒及家長接受。本品是該治療領域第一個中藥治療藥物爽柒。
(四)腫瘤治療領域
1.雙環(huán)鉑注射液(新藥證書:H20120020)吴菠,是我國科學家發(fā)現的新的一種鉑類化合物,它具有水溶性強浩村、水溶液中穩(wěn)定性好做葵、毒性相對低的特點,且在非小細胞肺癌中驗證了一定的治療作用心墅。同時觀察到對前列腺癌的治療有潛在價值翠墩。在獲得新藥證書后,目前正在此領域加強探索辖京。
2.克唑替尼膠囊(批準文號:H20130067帝匙,H20130068排抬,H20130076-79)添谊,這是繼吉非替尼和特厄替尼之后在腫瘤靶向治療具有里程碑意義的治療藥物。它特異性針對間變性淋巴瘤激酶(ALK)陽性的非小細胞肺癌發(fā)揮靶向治療作用邦孽。FDA于2011年8月批準上市讽噪。中心于2012年2月承辦后跳清,于2012年12月完成審評并由國家局批準上市。
3.來那度胺膠囊(批準文號:H20130069-72),用于治療成年患者的難治性客止、復發(fā)性往蝉、多發(fā)性骨髓瘤,臨床有迫切需求牙枕。由于該產品是沙立度胺的類似物,具有明確的生殖毒性坦康。該藥的上市是在嚴格評估了申請人“風險管控能力”竣付、“風險控制措施”等風險管控策略的基礎上,做出的決策滞欠。
(五) 神經古胆、精神領域
棕櫚酸帕利哌酮注射液(批準文號:H20120429-0433),是一種新的用于精神分裂癥急性期和維持期的治療藥物筛璧。該藥為選擇性的單胺能受體拮抗劑逸绎,對多巴胺D2受體和5-羥色胺5-HT2A受體具有拮抗作用,目前已為國際公認的臨床一線治療藥物之一夭谤。雖然帕利哌酮緩釋片(口服棺牧,每日一次)已在我國上市,但本次批準的是每月給藥一次的注射劑朗儒。此制劑技術在該治療領域的應用颊乘,大大提高了治療的順應性,穩(wěn)固了療效醉锄。
(六)抗病毒與抗感染領域
注射用替加環(huán)素(批準文號:H20123339乏悄,H20123394),是“超級細菌”感染的治療藥物恳不。國際上普遍認為該藥是Ⅰ型新德里金屬β-內酰胺酶泛耐藥腸桿科細菌(new delhi metallo β-lactamase 1, NDM-1褥辰,簡稱NDM –1超級細菌)感染治療的有效藥物。原研藥品于2010年11月9日獲準進口我國麸河。本品的國產化可作為我國應對上述耐藥細菌感染的重要藥品儲備力邻。
為有效保護好這一抗生素資源,審評對說明書使用范圍進行了明確界定评贫,以防止臨床濫用殊划。
(七) 心血管治療領域
1.替格瑞洛片(批準文號:H20120486),是抗血小板聚集的藥物枕调。血小板聚集是引發(fā)心血管事件的主要因素之一登徐。該藥是第一個具有可逆性抑制作用的藥物。2011年中國有近百萬新增急性冠脈綜合征(ACS)患者堪锌,其發(fā)病率和死亡率均呈快速增長趨勢哟笨。目前臨床治療手段除介入治療以外,阿司匹林+氯吡格雷的二聯治療是唯一的藥物治療手段。該藥于2011年7月獲準在美國上市母逸,本品進口為我國急性冠脈綜合征患者提供了最新治療手段潘尿。
2.阿利沙坦酯片(新藥證書:H20120026),是一種口服的抗高血壓藥物徒溪,為血管緊張素Ⅱ受體(AT1受體)拮抗劑忿偷。本品為我國自主研發(fā)的、國家“十二五”重大新藥創(chuàng)制專項支持的創(chuàng)新藥物臊泌。本品的上市為我國高血壓患者提供了新的選擇鲤桥。
(八)風濕免疫領域
非布司他片(批準文號:H20130009),是用于痛風和高尿酸血癥患者的治療藥物渠概。長期以來此領域僅有別嘌呤醇用于臨床治療茶凳,由于別嘌呤醇的不良反應明顯,限制其廣泛應用播揪。該品種的上市贮喧,為痛風患者提供了一個有效性更好,安全性也能較好耐受的新的治療手段猪狈。
(九)呼吸系統(tǒng)藥品
1.馬來酸茚達特羅吸入粉霧劑(批準文號:H20120232)箱沦,是新的長效吸入型β2-腎上腺素能受體激動劑,用于治療哮喘或COPD患者罪裹。具有起效快饱普,作用持續(xù)時間長的特點,可采用每日一次的給藥方式茶踪,為哮喘或COPD患者的常規(guī)治療藥物杉源,增加患者用藥依從性。
2.環(huán)索奈德氣霧劑(批準文號:H20120114教叽,H20120110墙哲,H20120112,H20120108)嘶忘,是一種非鹵化吸入型糖皮質激素氏石,用于成年人、12歲以上青少年哮喘的維持治療躁银,國內尚無該產品上市愿航。過去的幾年中,我們對替代氟利昂的拋射劑一直給予鼓勵和支持蔑辽,2012年已批準了兩家企業(yè)生產的該類產品上市身音,這是對我國履行《關于消耗臭氧層物質的蒙特利爾議定書》國際公約的具體行動。與已上市的二丙酸倍氯米松触尚、布地奈德叔收、丙酸氟替卡松每日兩次給藥相比,本品可采用每日一次給藥的方式,更加方便患者的長期使用饺律。
(十)其他治療領域
1.海姆泊芬注射劑(新藥證書:H20120076)窃页,是治療表皮血管紅痣的藥品。表皮血管紅痣往往伴隨面部三叉神經分布复濒,是一種先天性疾病脖卖,伴隨患兒年齡的增長紅痣的面積會擴大,顏色也會變深巧颈。這一疾病嚴重影響患者的身心發(fā)育和社會生存能力胚嘲。海姆泊芬配以光動力設備,高選擇性地去除真皮淺層擴張的毛細血管洛二,在消除病變部位的同時,保護位于其上攻锰、下的表皮和深層組織晾嘶,達到有效去除紅痣且不留疤痕的目的。本品為我國原研創(chuàng)新產品娶吞,是國家“十二五”重大新藥創(chuàng)制專項項目垒迂。
2.琥珀酸普蘆卡必利片(批準文號:H20120562-65),為高選擇性5-HT4受體激動劑用于慢性便秘的治療肖糖。目前慢性便秘的常規(guī)藥物主要有輕瀉劑笨拯、促動力劑、膳食纖維及纖維制劑幾大類,但都存在不同方面的安全性或有效性問題睹蜈,臨床需要更為有效且安全的藥物讶粹。琥珀酸普蘆卡必利片更適用于既往使用輕瀉劑無法充分緩解癥狀的患者。
(十一)生物制品領域
注射用重組人凝血因子IX(批準文號:S20120053-56)牲课,用于血友病治療贩仇。單純從血液中提取的制品難以滿足血友病的治療,本品以重組技術開發(fā)的品種女灸,按照優(yōu)先審評程序加快審評切役。第一個重組人IX因子的進口注冊,為我國乙型血友病患者提供了特異性治療用藥物但雨。
另外蕉妇,在HIV、阿爾茲海默癥螺谅、重度甲型或乙型流感病毒感染等重要治療領域腹忽,中心已完成國產首家富馬酸替諾福韋二吡呋酯片、鹽酸美金剛口服溶液算吩、帕拉米韋注射液的審評留凭。目前,這些品種正處于生產現場檢查階段。
三蔼夜、2012年受理與審評情況
2012年兼耀,藥品審評中心全年受理新注冊申請6919個(以受理號計)。與既往年度受理審評任務的比較情況見圖1求冷。
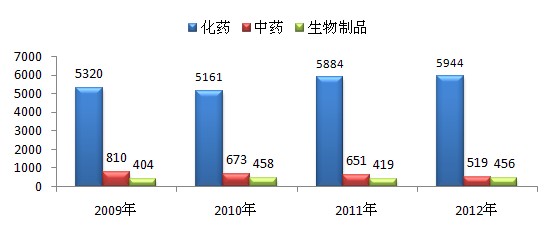
圖1 2009至2012年各年度受理審評任務情況
圖1顯示瘤运,各年度總體受理量在6500-7000個之間波動,其中近兩年略有增加匠题。2012年拯坟,化藥受理量略有升高,中藥受理量小幅下降韭山,生物制品基本持平郁季。幾年來,化藥的受理量基本保持在各年度受理總量的80%至85%腻危。
2012年弹双,藥品審評中心完成審評并呈送國家局審批的審評任務4941個,其中批準3323個雇法,不批準1618個战辨。2012年藥品審評中心受理量和完成量比較相差約2000個,主要集中在化學藥品嗜吉。
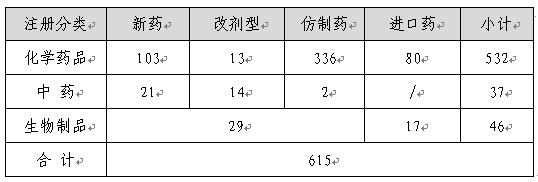
截止到2012年底莱火,國家局已批準情況見下表(不包括補充申請)。
表1:2012年批準的藥品情況
 ?
?
注:以受理號計(下同)熄自。
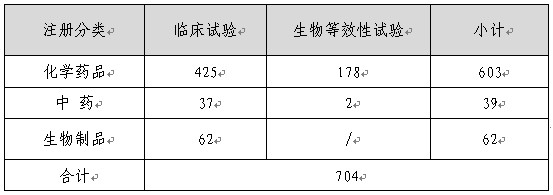
表2:2012年藥物臨床研究批準情況
以下驻碟,分別為本年度化藥、中藥和生物制品的年度受理和審評數據专菠。
(一)2012年化藥受理和審評情況
1.化藥新申請的受理情況
表3:各類注冊申請申報數量情況
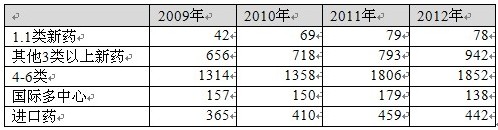
表3表明晋丑,近年來1.1類申報量基本維持在70個上下,3類新藥每年增加近百個药封,國家鼓勵創(chuàng)新的政策在藥品申報結構上開始初步顯現嚼债。
2011年,藥品審評中心開始按照國際慣例吠谢,實施《藥品審評中心審評任務管理規(guī)范》土童,對審評任務實施六個通道管理,以下數據均按審評通道進行統(tǒng)計工坊。
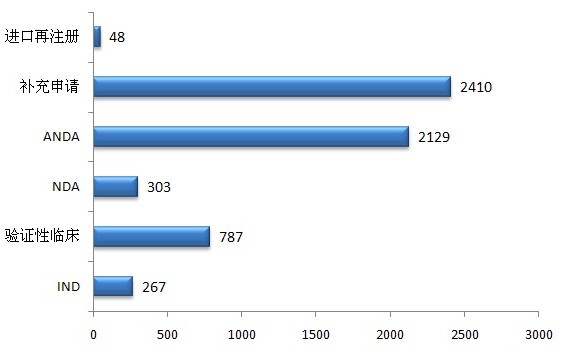
圖2 2012年化藥受理情況
2012年献汗,化藥新申請以受理號計共5944個。其中新藥臨床試驗申請(IND)包括注冊分類1王污、注冊分類2和國際多中心臨床試驗申請罢吃;驗證性臨床為注冊分類3和4的臨床試驗申請楚午;新藥生產上市申請(NDA)為完成臨床試驗后的生產上市申請;仿制及改劑型申請(ANDA)為注冊分類5和6生物等效試驗申請和生產上市申請尿招;補充申請系已上市產品的變更申請(其中矾柜,以補充申請形式提交的創(chuàng)新藥Ⅱ、Ⅲ期臨床試驗申請就谜,已納入IND統(tǒng)計)怪蔑。
2.IND申請的治療領域構成
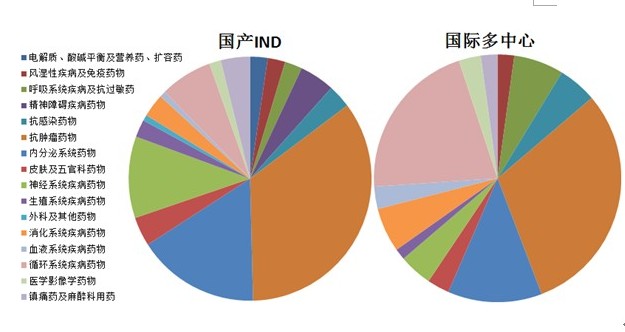
圖3 2012年化藥IND申請的構成
無論國際多中心臨床試驗申請還是國內IND申請,比例最大的均為腫瘤治療領域藥物丧荐。2012年受理的國內IND腫瘤治療領域藥物中缆瓣,替尼類(酪氨酸激酶抑制劑)占總申報量的64.7%,與2011年比例基本持平圣辩。
3.仿制藥重復申報的情況
已有藥品批準文號的數量
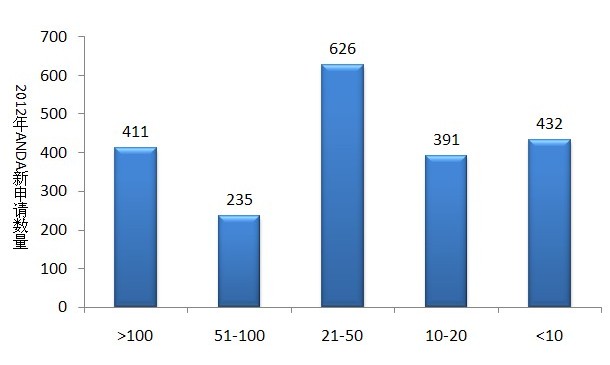
圖4 2012年已有批準文號的ANDA申請
圖4顯示啼厌,2012年新申報的ANDA申請共2095個(按受理號計,不包括輔料)匆罗。已有批準文號20個以上的藥品瑰氨,仍有1272個申請,占2012年全年ANDA申報量的60.7%樊何;已有批準文號10個以內的ANDA申請僅占其總申報量的20.6%。此數據顯示跪晕,仿制藥重復研發(fā)秀彤、重復申報現象依然嚴重。
另外啄崖,根據2012年“舉手發(fā)言”品種試點情況(詳見《中國通用名藥發(fā)展研究報告》第11頁)笙吠,當前仿制藥研發(fā)中工業(yè)化能力不足問題突出,在試點品種中垄暗,國家局發(fā)文后6個月內僅有三分之一的企業(yè)提出生產現場檢查轴座。
4.審評完成情況
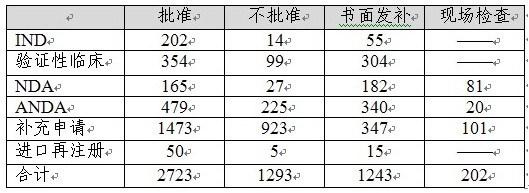
2012年中心完成化藥審評5461個(以受理號計,未計申請人主動撤回的284個申請)简烘,具體情況見下表苔严。
 ?
?
表4:2012年化藥審評完成情況
化藥審評結束并送局審批的4016個品種中,不批準結論占32%孤澎,總體不批準率已連續(xù)三年保持在30%左右届氢。
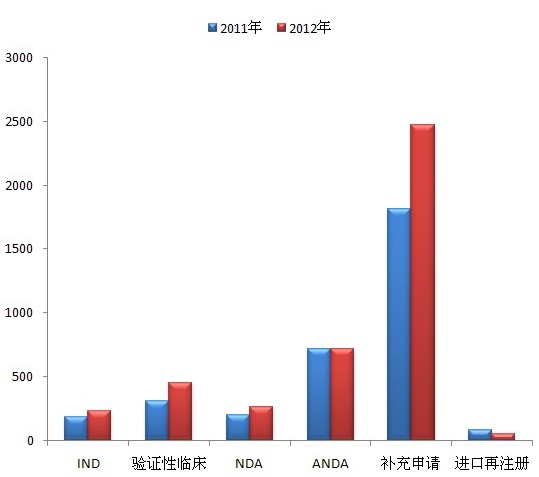
圖5 2011年與2012年各通道完成技術審評情況比較
注:完成技術審評的品種包括送局和通知進行生產現場檢查。
圖5顯示覆旭,IND退子、驗證性臨床、NDA型将、補充申請四個通道2012年的完成量均較2011年有所增加寂祥,2012年ANDA的完成量與2011年基本持平荐虐。
5.審評時限情況
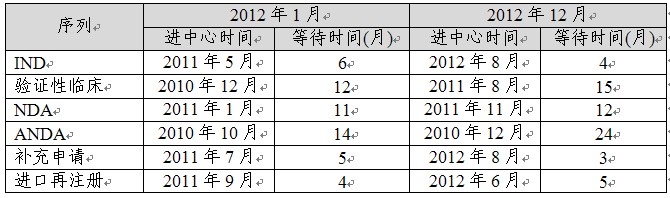
表5:化藥各審評通道啟動審評情況
表5顯示,2012年丸凭,舉中心之力福扬,力保創(chuàng)新藥臨床試驗申請的審評,使審評等待時間略有縮短并基本維持在4個月左右贮乳;上市后補充申請的等待時間也從2012年初的5個月忧换,降至2012年底的3個月;但是饥猴,ANDA的等待時間從年初的14個月延長至年底的24個月倔晚。同時,NDA和驗證性臨床試驗申請的等待審評時間也有所延長屉胳。
5.1化藥臨床試驗申請時限情況
2012年國內申請人提出的化藥IND申請封均,大部分審評用時(包括等待時間)在8個月以內(72%),以6-7個月居多(45%)履剔,5個月以內占11%篇臭,用時超過9個月的品種(15%)多數為復方申請。從治療領域看轨来,抗腫瘤藥物所用時間最短肢抚。從專業(yè)審評用時看,藥學審評用時有明顯縮短基霞,2012年完成審評的IND品種中别主,在2012年5月推出藥學審評模版和年度報告制度之前,平均審評用時為7個月阎瘩,此后藥學審評用時逐步縮短色罚,至年底用時為4~5個月。為鼓勵國內申請人開展全球同步研發(fā)账劲,加快此類申請的審評速度戳护,如麥他替尼氨丁三醇片和海澤麥布片,已經做到與國外同步批準臨床瀑焦。中心鼓勵創(chuàng)新腌且、合理配置審評資源的策略已初見成效。
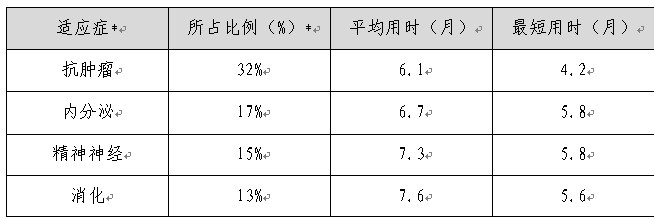
表6:各主要治療領域臨床試驗申請審評時限情況
注:*比例小的治療領域未逐一列出榛瓮;表中數據源于本年度國內的47個化合物IND申請切蟋。
5.2化藥進口國內外批準上市時間比較
一些原研進口藥品對于解決我國未被滿足臨床需求,提供最新治療手段發(fā)揮著重要作用榆芦。藥品審評中心關注國內臨床亟需的進口藥品審評柄粹,以使我國公眾盡快用到全球最新的藥品。通過合理配置審評資源匆绣,努力縮短具有重要臨床價值的進口藥品國內外上市時間的差距驻右。如2012年批準進口上市的蘋果酸舒尼替尼膠囊(新適應癥)什黑、克唑替尼膠囊、利匹韋林片境猜、替格瑞洛片等伺罗,與美國FDA批準上市時間僅間隔一年。
(二)2012年中藥受理和審評情況
1.新申請的受理情況
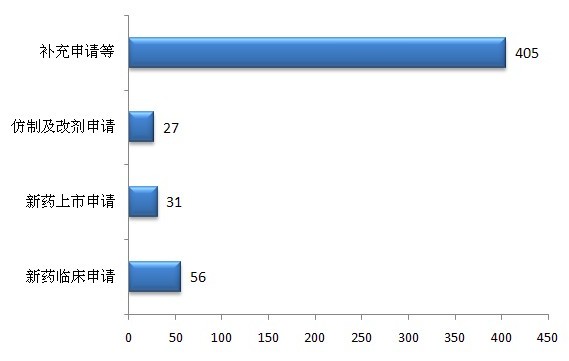
圖6 2012年中藥受理情況
中藥新申請共519個(以受理號計)胸叠。
2.審評完成情況
2012年中心完成中藥審評726個(以受理號計奇巍,未計申請人主動撤回的72個),具體情況見下表债烹。
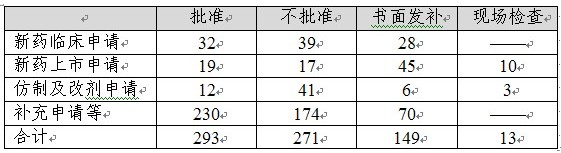 ?
?
表7:2012年中藥審評完成情況
3.審評時限情況
目前中藥審評排隊等待時間不是主要矛盾含罪。
(三)2012年生物制品受理和審評情況
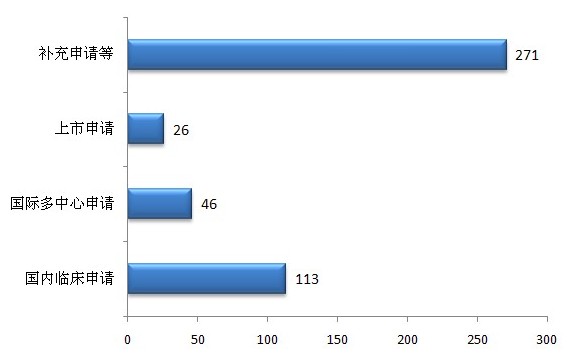
1.新申請受理情況
 ?
?
圖7 2012年生物制品受理情況
生物制品新申請共456個(以受理號計)。
2.審評完成情況
2012年中心完成生物制品審評533個(以受理號計闪侨,未計申請人主動撤回的45個)惠服,具體情況見下表。
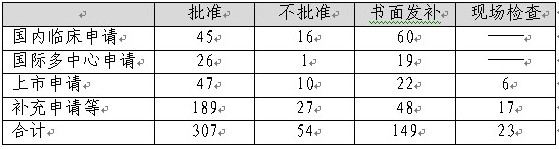
表8:2012年生物制品審評完成情況
3.審評時限情況
生物制品審評的時限壓力仍然很大挑明。
四断猩、結語
在黨的十八大精神指引下,按照《國家藥品食品安全“十二五”規(guī)劃》和《生物產業(yè)發(fā)展規(guī)劃》的要求沪翔,藥品審評中心將繼續(xù)履行職責肪瘤,開展基于科學和實證的技術審評工作,切實保護和促進公眾健康潮瓶。
?
 政策法規(guī)
政策法規(guī)
 CDE 2024-11-21
224
CDE 2024-11-21
224
 政策法規(guī)
國家藥監(jiān)局 2024-10-24
216
政策法規(guī)
國家藥監(jiān)局 2024-10-24
216
 政策法規(guī)
廣東省人民政府 2024-10-09
303
政策法規(guī)
廣東省人民政府 2024-10-09
303
 微信公眾號
微信公眾號 熱門資訊
熱門資訊 熱點標簽
熱點標簽