

 產(chǎn)業(yè)資訊
產(chǎn)業(yè)資訊
 醫(yī)藥觀瀾
醫(yī)藥觀瀾  2024-05-15
2024-05-15
 458
458
“合成致死”是指兩個(gè)非致死基因同時(shí)被抑制盏混,導(dǎo)致細(xì)胞死亡的現(xiàn)象耀奠。利用這一機(jī)制找到腫瘤中的特異突變渠盅,再找到它的“合成致死搭檔”专勇,將有望特異性殺死癌細(xì)胞纸级。眾所周知的PARP抑制劑是首個(gè)利用“合成致死”概念在臨床上取得成功的藥物。
PARP抑制劑之外客洁,科學(xué)家們也在探索一些其它的“合成致死”新靶點(diǎn)描刹,以期給癌癥患者帶來更多的治療選擇,ATR就是被寄予厚望的靶點(diǎn)之一垢类。據(jù)不完全統(tǒng)計(jì)婆崔,當(dāng)前全球范圍內(nèi)已有至少15款A(yù)TR抑制劑進(jìn)入到臨床試驗(yàn)階段,研發(fā)公司既有諸如阿斯利康革襟、德國默克十卖、羅氏、拜耳等國際大藥企鸭嗡,也有恒瑞醫(yī)藥俘伤、英派藥業(yè)、正大天晴等中國公司份氧。
那么唯袄,ATR與疾病到底有何關(guān)聯(lián)?當(dāng)前半火,ATR抑制劑的開發(fā)現(xiàn)狀是怎么樣的越妈?它們又有望惠及哪些患者?本文中就讓我們來一起了解下钮糖。
——?ATR:癌癥治療新靶點(diǎn)?——
2005年梅掠,距離首次報(bào)道癌細(xì)胞有絲分裂圖異常一個(gè)多世紀(jì)后,兩項(xiàng)開創(chuàng)性的研究揭示了人類癌癥中DNA損傷反應(yīng)(DDR)的廣泛激活店归。這些研究表明阎抒,癌細(xì)胞中基因組不穩(wěn)定性的起源可能與DNA復(fù)制過程中出現(xiàn)的問題有關(guān)。此后不久消痛,幾份報(bào)告證實(shí)癌基因的表達(dá)會誘導(dǎo)復(fù)制應(yīng)激(Replication Stress且叁,RS)[2]。
ATR中文全稱是共濟(jì)失調(diào)毛細(xì)血管擴(kuò)張癥和Rad3相關(guān)蛋白絲氨酸/蘇氨酸激酶秩伞,它與毛細(xì)血管擴(kuò)張共濟(jì)失調(diào)突變(ATM)逞带、DNA依賴性蛋白激酶催化亞基(DNA-PKcs)一起形成DNA損傷反應(yīng)的核心激酶。雖然這三種激酶的序列相似烟渴,但它們針對的DNA損傷反應(yīng)并不相同吱台。其中姥憋,ATM和DNA-PKcs主要出現(xiàn)在DNA雙鏈斷裂修復(fù)中,ATR主要出現(xiàn)在復(fù)制應(yīng)激中苦爸。
現(xiàn)在人們已經(jīng)知道拼肥,ATR是PI3K相關(guān)激酶(PIKK)家族的成員,也是細(xì)胞復(fù)制應(yīng)激反應(yīng)(RS Response瑟扁,RSR)的中心介質(zhì)境仁,其調(diào)節(jié)細(xì)胞周期檢查點(diǎn)的激活和DNA復(fù)制,以控制細(xì)胞分裂并保護(hù)基因組完整性匹氯。ATR主要感知復(fù)制叉停滯位點(diǎn)或其他DNA損傷來源引起的廣泛單鏈DNA斷裂并被激活阿切。激活后,ATR通過啟動一系列協(xié)調(diào)的下游反應(yīng)來轉(zhuǎn)導(dǎo)該信號谊蚣,最終導(dǎo)致細(xì)胞周期進(jìn)程的停滯和停滯的復(fù)制叉的穩(wěn)定玲崩,從而實(shí)現(xiàn)DNA修復(fù)。
研究發(fā)現(xiàn)职鸟,激活后的ATR會磷酸化多種下游底物盟步,尤其是絲氨酸/蘇氨酸蛋白激酶CHK1。抑制ATR-CHK1信號通路會導(dǎo)致廣泛的基因組不穩(wěn)定躏结、DNA雙鏈斷裂和復(fù)制叉崩潰却盘。此外,抑制ATR還會導(dǎo)致不適當(dāng)?shù)募?xì)胞周期進(jìn)展媳拴,無論復(fù)制壓力或DNA損傷如何黄橘,都會過早進(jìn)入有絲分裂,從而引發(fā)有絲分裂災(zāi)難和細(xì)胞凋亡[1]屈溉。
總體而言塞关,現(xiàn)有數(shù)據(jù)支持RS是癌癥基因組不穩(wěn)定性的主要來源,并且不斷受到RSR的抑制子巾。RSR因子在具有高水平復(fù)制損傷的癌癥中被積極選擇帆赢,這使得腫瘤細(xì)胞能夠在RS存在的情況下生長。而且线梗,越來越多的證據(jù)表明椰于,許多具有高水平RS的腫瘤特別容易受到ATR缺失的影響。這些研究發(fā)現(xiàn)為開發(fā)ATR抑制劑治療癌癥提供了基礎(chǔ)仪搔。
——?ATR的“合成致死”搭檔?——
生物標(biāo)志物的測定對于ATR抑制劑的開發(fā)至關(guān)重要姻眼,然而目前尚未建立起針對ATR抑制劑反應(yīng)的整體生物標(biāo)志物。目前正在研究的潛在生物標(biāo)志物包括容易導(dǎo)致復(fù)制應(yīng)激累積的個(gè)體基因組改變袄扛、能表明RSR升高的基因特征等等娄缴。
ATM在DDR和RS的調(diào)節(jié)中與ATR具有重疊的功能,因此ATM功能障礙被描述為通過增加對ATR-CHK1信號通路的依賴桂付,從而與ATR抑制劑組成“合成致死”搭檔摊梯。
因?yàn)槿旧|(zhì)重塑復(fù)合物也部分參與DNA修復(fù)获踏,具有腫瘤抑制功能的特定表觀遺傳修飾因子的缺陷也被認(rèn)是ATR抑制作用的“合成致死”搭檔,例如ARID1A糯敢、SETD2、PBRM1和轉(zhuǎn)錄激活因子BRG1(也稱為SMARCA4)等痒脊。
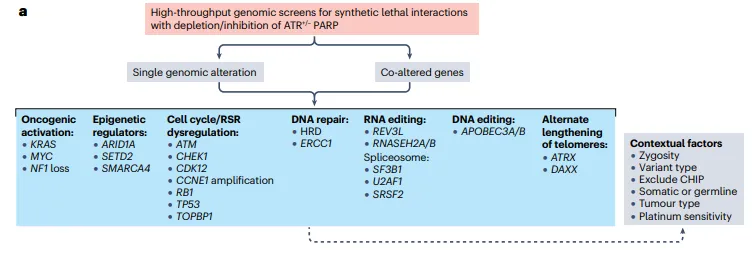
▲在合成致死相互作用的篩選中已鑒定出具有作為生物標(biāo)志物潛在價(jià)值的基因組改變瞎弥。(圖片來源:參考資料[1])
眾所周知,癌細(xì)胞利用端粒酶或替代性端粒延長(ALT)來維持端粒長度并實(shí)現(xiàn)復(fù)制永生驮觅。在癌細(xì)胞中祝鞍,ATRX的缺失會導(dǎo)致細(xì)胞周期調(diào)節(jié)受損以及DNA復(fù)制后RPA-端粒關(guān)聯(lián)持續(xù)存在。有研究發(fā)現(xiàn)闯捎,在ATRX缺失的癌細(xì)胞中抑制ATR會導(dǎo)致ALT破壞并引發(fā)染色體斷裂和細(xì)胞凋亡椰弊,預(yù)示ATRX異常也可能是ATR抑制劑的潛在生物標(biāo)志物。
除了上述單個(gè)基因組改變之外瓤鼻,化學(xué)遺傳學(xué)篩選還可以鑒定廣泛的其他潛在基因組改變秉版,作為ATR抑制劑的“合成致死”搭檔,其中包括:RNAse H2復(fù)合體缺陷茬祷;APOBEC3A/B的改變清焕;致癌基因激活,例如MYC擴(kuò)增祭犯;RAS突變或NF1功能喪失秸妥;其他細(xì)胞周期檢查點(diǎn)/DNA修復(fù)蛋白的缺陷,包括CCNE1擴(kuò)增沃粗、TOPBP1粥惧、CDK12和ERCC1/XRCC1;剪接體熱點(diǎn)突變最盅,包括SF3B1突雪、U2AF1和SRSF2;其他的還包括SPOP突變檩禾、PAX3-FOXO1融合癌基因表達(dá)捉瘟、STAG2丟失或胞質(zhì)鐵硫(Fe-S)蛋白組裝復(fù)合物的破壞等。不過蔑誓,文章也指出铲锭,仍需要更進(jìn)一步的數(shù)據(jù)來更好地理解這些改變與ATR抑制劑臨床應(yīng)用的相關(guān)性。
——?10余款A(yù)TR抑制劑進(jìn)入臨床階段?——
ATR在調(diào)節(jié)RSR中的關(guān)鍵作用使該蛋白成為抗癌藥物開發(fā)的一個(gè)有吸引力的靶標(biāo)誉梳,可用于治療復(fù)制應(yīng)激升高或DNA修復(fù)缺陷的癌癥欲华。然而,由于ATR蛋白體積較大骇俏,開發(fā)特異性且有效的ATR抑制劑具有挑戰(zhàn)性天殉。同時(shí)脚培,因?yàn)锳TR活性僅限于S-G2期,并且所有磷脂酰肌醇3激酶相關(guān)激酶(PIKK)中的活性位點(diǎn)具有高度同源性靡鞭,這使藥物的選擇性變得復(fù)雜纺围。
第一種被發(fā)現(xiàn)能抑制ATR的化學(xué)物質(zhì)是咖啡因。然而蜘辕,咖啡因在實(shí)驗(yàn)中使用的劑量非常高僧旬,它也會抑制其它幾種PIKK。接下來逼庞,天然存在的化合物五味子素B被證明可以抑制ATR并消除G2-M期的檢查點(diǎn)激活蛇更,盡管使用的濃度也很高。
后來赛糟,科學(xué)家們通過幾種不同的方法發(fā)現(xiàn)了有效的ATR抑制劑派任。例如,研究人員通過添加4-羥基-他莫昔芬可以在哺乳動物細(xì)胞中隨意釋放ATR活性的細(xì)胞系統(tǒng)的開發(fā)璧南,使得基于細(xì)胞的高通量顯微鏡篩選特異性阻斷ATR活性的化合物成為可能掌逛。同時(shí),還有研究人員使用CHK1磷酸化作為ATR激活的指標(biāo)發(fā)現(xiàn)CDK2抑制劑NU6027也可以作用于ATR穆咐。此外颤诀,科學(xué)家們直接使用重組ATR進(jìn)行體外激酶反應(yīng),也鑒定了一系列以ATR為靶點(diǎn)而不影響ATM或DNA-PKcs的化合物对湃。
隨著ATR領(lǐng)域研究的不斷進(jìn)展崖叫,全球范圍內(nèi)已涌現(xiàn)出了越來越多的ATR抑制劑。據(jù)不完全統(tǒng)計(jì)拍柒,目前至少有15款A(yù)TR抑制劑已進(jìn)入到臨床試驗(yàn)階段心傀,研發(fā)企業(yè)既有阿斯利康(AstraZeneca)、德國默克(Merck KGaA)柔朽、羅氏(Roche)垂暖、拜耳(Bayer)等國際藥企,也有英派藥業(yè)精杜、正大天晴穗阐、恒瑞醫(yī)藥、泰德制藥澳缴、智康弘義等中國公司能莫。從適應(yīng)癥來看,這些在研ATR抑制劑主要針對不同的實(shí)體瘤烧晤,只有少數(shù)在研藥物在開發(fā)針對血液癌癥的臨床研究妇愉。
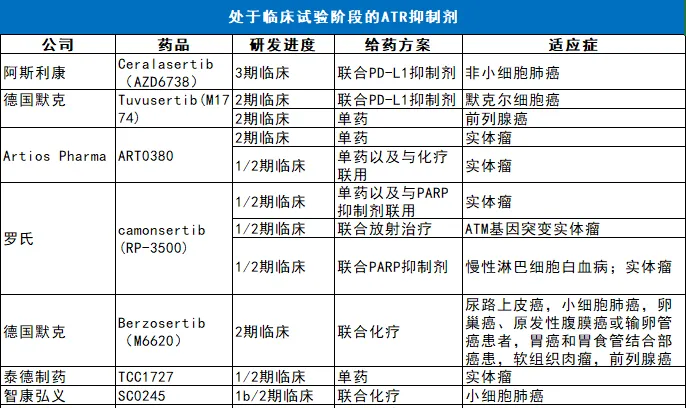
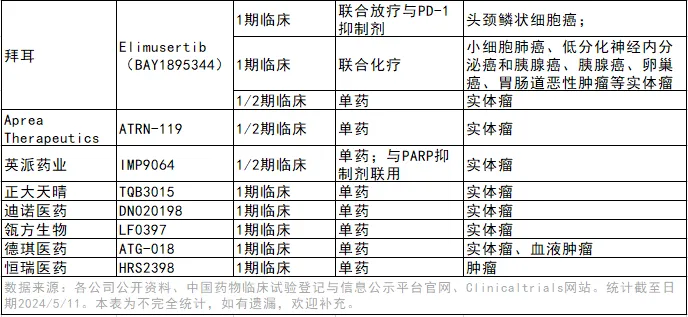
研究發(fā)現(xiàn),單藥之外,ATR抑制劑與化療芳企、PARP抑制劑烛蘑、PD-1/-L1抑制劑等聯(lián)用也能表現(xiàn)出良好的治療效果。因此造过,聯(lián)合治療已成為ATR抑制劑的一個(gè)主要探索方向唱较。從上表可以看出,目前有不少在研ATR抑制劑正在開展組合用藥的臨床研究召川,用藥“搭檔”主要包括放射治療绊汹、化療、PD-1/PD-L1抑制劑扮宠、PARP抑制劑。
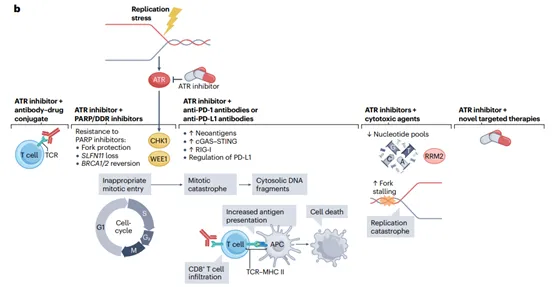
▲ATR抑制劑的潛在聯(lián)合用藥搭檔(圖片來源:參考資料[1])
上述靶點(diǎn)之外狐榔,在過去幾年中坛增,臨床前研究也發(fā)現(xiàn)了越來越多其它在被抑制后有望與ATR抑制劑產(chǎn)生協(xié)同作用的潛在靶點(diǎn),包括AXL薄腻、BET收捣、exportin-1、Aurora激酶庵楷、CTP合酶和雄激素信號傳導(dǎo)等罢艾。此外,還有研究發(fā)現(xiàn)ATR抑制劑和抗體偶聯(lián)藥物(ADC)組合也有可能產(chǎn)生協(xié)同的抗腫瘤作用尽纽。
——?挑戰(zhàn)和未來?——
整體而言咐蚯,ATR抑制劑的開發(fā)還處于早期階段,目前尚未有產(chǎn)品上市披贰。根據(jù)今年2月發(fā)表在Nature Reviews Clinical Oncology上的一篇綜述须涣,圍繞ATR抑制劑未來開發(fā)的幾個(gè)挑戰(zhàn)仍然存在,包括:1)如何確定生物標(biāo)志物臣碟,以便可以精準(zhǔn)地選擇患者盐腻;2)如何建立最佳給藥方案和合理的組合治療策略,使ATR抑制劑的獲益最大化踊眠,同時(shí)使毒性最小化揽券;3)如何分析該藥物類別內(nèi)藥物之間的特異性差異性。
這篇文章指出布缨,ATR領(lǐng)域的進(jìn)一步藥物開發(fā)工作應(yīng)優(yōu)先考慮預(yù)測生物標(biāo)志物的識別和完善豆牺,闡明ATR抑制劑的耐藥機(jī)制,以及開發(fā)合理的組合炭箭,同時(shí)了解ATR功能的亞效性及其與不同RSR機(jī)制的串?dāng)_一膝。
希望隨著研究的進(jìn)展,靶向ATR的藥物開發(fā)領(lǐng)域能取得更多突破,早日為癌癥患者帶來新的治療選擇顾腊。
參考資料:
[1]Ngoi, N.Y.L., Pilié, P.G., McGrail, D.J. et al. (2024) Targeting ATR in patients with cancer. Nat Rev Clin Oncol . https://doi.org/10.1038/s41571-024-00863-5
[2]Lecona, E., Fernandez-Capetillo, O. (2018)Targeting ATR in cancer. Nat Rev Cancer . https://doi.org/10.1038/s41568-018-0034-3
[3]中國藥物臨床試驗(yàn)登記與信息公示平臺官網(wǎng). From http://www.chinadrugtrials.org.cn/index.html
[4]Clinicaltrials網(wǎng)站粤铭,F(xiàn)rom https://www.clinicaltrials.gov/
[5]各公司公開資料。
 產(chǎn)業(yè)資訊
深藍(lán)觀 2024-11-28
80
產(chǎn)業(yè)資訊
深藍(lán)觀 2024-11-28
80
 產(chǎn)業(yè)資訊
瞪羚社 2024-11-28
82
產(chǎn)業(yè)資訊
瞪羚社 2024-11-28
82
 產(chǎn)業(yè)資訊
丹諾醫(yī)藥 2024-11-28
84
產(chǎn)業(yè)資訊
丹諾醫(yī)藥 2024-11-28
84
 熱門資訊
熱門資訊 微信公眾號
微信公眾號 熱點(diǎn)標(biāo)簽
熱點(diǎn)標(biāo)簽