

 產(chǎn)業(yè)資訊
產(chǎn)業(yè)資訊
 藥渡
藥渡  2024-07-04
2024-07-04
 336
336
FDA對于罕見病基因療法的監(jiān)管靈活性和尺度把握,已經(jīng)成為了行業(yè)關(guān)注的焦點(diǎn)話題喊积。而這個(gè)話題的熱度烹困,將在今年持續(xù)增溫。兩款未實(shí)現(xiàn)主要終點(diǎn)的DMD基因療法和Elevidys注服,為FDA的罕見病基因療法審批靈活性提供了兩道難題。
DMD療法Viltepso驗(yàn)證性試驗(yàn)失敗
日本制藥商N(yùn)ippon Shinyaku日前表示措近,他們的杜氏肌營養(yǎng)不良癥(DMD)藥物Viltepso在一項(xiàng)安慰劑對照驗(yàn)證性試驗(yàn)中未能達(dá)到其主要終點(diǎn)。研究結(jié)束時(shí)瞭郑,接受藥物治療的兒童可以更快地從地板上站起來辜御,但接受安慰劑的兒童也能從地板上站起來。各組之間沒有統(tǒng)計(jì)學(xué)上的顯著差異屈张。Viltepso在2020年8月12日得到了FDA的加速批準(zhǔn)。
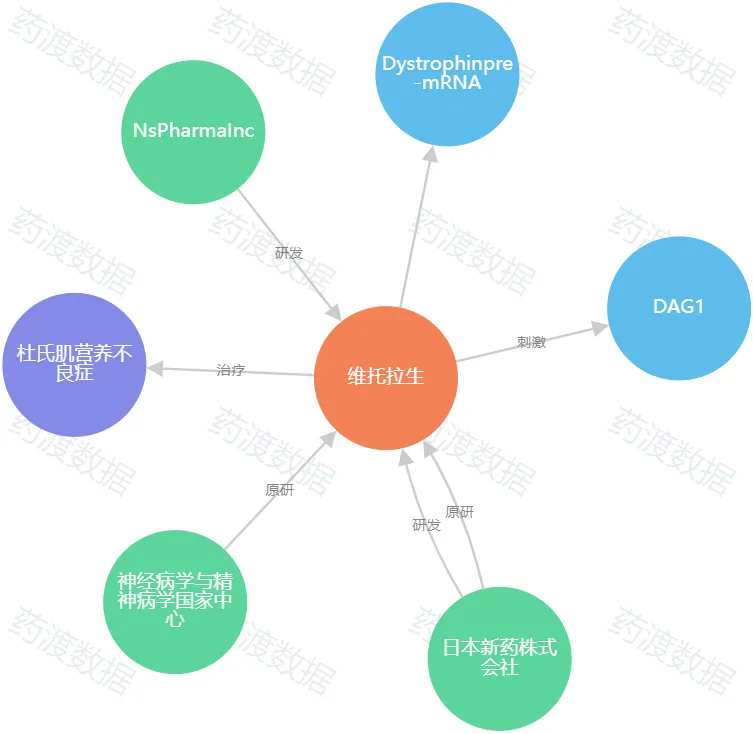
圖1.Viltolarsen知識(shí)圖譜(圖源:藥渡數(shù)據(jù))
盡管Nippon沒有公布詳細(xì)數(shù)據(jù)讽益,但這次試驗(yàn)失敗卻尤其值得注意逊床。這是因?yàn)樵囼?yàn)藥物Viltepso屬于存在爭議的exon-skipper類型。DMD是由一種dystrophin基因的突變所導(dǎo)致dystrophin蛋白生成缺陷為病理的联喘。Exon-skipper類型的藥物目的在于增加患者的dystrophin蛋白數(shù)量珊蟀。但此類藥物只會(huì)略微增加dystrophin蛋白的水平,在臨床益處方面未能得到大型安慰劑對照研究的證實(shí)人杜。
Viltepso失敗帶給DMD療法開發(fā)的影響
Sarepta于2016年獲得了Exon-skipper藥物Exondys 51(eteplirsen)的監(jiān)管批準(zhǔn)。在其背后坷疙,DMD患兒家長的激烈游說活動(dòng)為其提供了重要的支持皱蝙。FDA要求Sarepta在2020年前完成Exondys 51的上市后驗(yàn)證性研究,但該研究直到2020年才開始矮层,目前尚未完成,不過這并不妨礙Sarepta在接下來的時(shí)間內(nèi)上市兩款Exon-skipper藥物并獲得數(shù)十億美元的收入锌烫。
Nippon Shinyaku的數(shù)據(jù)加劇了DMD療法批準(zhǔn)標(biāo)準(zhǔn)的爭論殉俗。但從目前的情況來看圾纤,這次試驗(yàn)失敗不太可能顯著改變FDA批準(zhǔn)此類藥物的態(tài)度。Viltepso的研究涉及77名患者倦逐,針對exon 53,而不是Exondys 51中exon 51的檬姥。這項(xiàng)試驗(yàn)只持續(xù)了48周曾我,試驗(yàn)失敗的原因也可能于時(shí)間過短有關(guān),無法顯示療效在那些進(jìn)展緩慢的適應(yīng)癥中的差異健民。
Sarepta針對Vyondys 53(golodirsen抒巢,針對exon 53)和Amondys 45(Casimersen秉犹,針對exon 45)的隨機(jī)三期試驗(yàn)已經(jīng)開始蛉谜,為期兩年,這兩款Exon-skipper DMD療法都是在Exondys 51之后獲得加速批準(zhǔn)上市的型诚。針對Exondys 51的上市后驗(yàn)證性研究計(jì)劃為期三年。
對于DMD試驗(yàn)終點(diǎn)的討論也是這個(gè)領(lǐng)域內(nèi)的焦點(diǎn)鸳劳。以上的驗(yàn)證性試驗(yàn)采取了起床時(shí)間或六分鐘步行測試等作為試驗(yàn)終點(diǎn)狰贯,而患者利益和倡導(dǎo)者群體則強(qiáng)調(diào)受試療法更微妙的益處。現(xiàn)已退休的FDA高級官員Janet Woodcock參與了Exondys 51的批準(zhǔn)念澜,她認(rèn)為大多數(shù)DMD患者在青少年時(shí)期就必須坐在輪椅上,這會(huì)導(dǎo)致壽命縮短至30歲左右禾样。但如果他們?nèi)匀豢梢砸苿?dòng)電腦鼠標(biāo)并進(jìn)行交流驱香,那將產(chǎn)生巨大的影響。
盡管驗(yàn)證性試驗(yàn)失敗怨瑰,但Nippon Shinyaku在其公告中簡稱摇昌,他們?nèi)匀幌嘈牌溆赩iltepso是“有益的”,并引用了使用Viltepso約四年的患者的觀察數(shù)據(jù)篓围。Nippon Shinyaku表示將與FDA商討這款藥物的未來。
無論如何培按,Viltepso成為了第二個(gè)大型安慰劑對照試驗(yàn)失敗的Exon-skipper DMD療法案例嘉警。2013年,葛蘭素史克Exon 51藥物的3期研究也未能達(dá)到其主要終點(diǎn)沪识。今年年底可能會(huì)有更多數(shù)據(jù)公布拼缝,屆時(shí)Sarepta的Exondys 51的3期試驗(yàn)結(jié)果將出爐彰亥。Vyondys 53(golodirsen)和Amondys 45(Casimersen)的3期數(shù)據(jù)可能將于2025年問世咧七。
FDA批準(zhǔn)了臨床失敗的Elevidys
Viltepso的上市后驗(yàn)證性試驗(yàn)的失敗再次為FDA制造了一個(gè)難題,是否將撤回這款DMD藥物的加速批準(zhǔn)继阻。而這個(gè)問題出現(xiàn)的時(shí)間節(jié)點(diǎn)恰好在FDA為頗受爭議的Sarepta DMD基因療法Elevidys做監(jiān)管決定的前夕耻涛。Sarepta的DMD基因療法Elevidys在2023年6月27日頂著巨大的爭議獲得了FDA的加速批準(zhǔn),CBER主任Peter Marks的決定對Elevidys的命運(yùn)產(chǎn)生了決定性的作用瘟檩。當(dāng)時(shí)FDA內(nèi)的意見是直接拒絕Elevidys的BLA申請抹缕,但最終這款藥物卻在Peter Marks的支持下,在咨詢委員會(huì)會(huì)議之后獲得了FDA的加速批準(zhǔn)決定卓研。
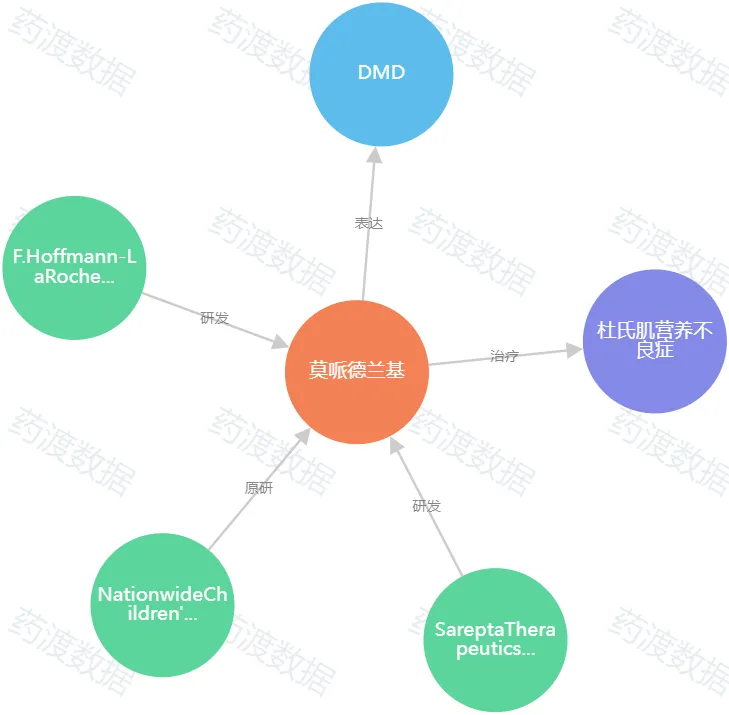
圖2.Elevidys知識(shí)圖譜(圖源:藥渡數(shù)據(jù))
盡管Elevidys在加速批準(zhǔn)后的III期上市后驗(yàn)證性研究中未能達(dá)到主要終點(diǎn),但從Elevidys的PDUFA日期之前各方預(yù)測來看睹簇,大家普遍看好FDA將加速批準(zhǔn)升級為全面批準(zhǔn)奏赘。原因之一在于FDA并沒有召開專家咨詢委員會(huì)會(huì)議,去年的該會(huì)議Elevidys獲得了微弱的投票贊成太惠。另一個(gè)重要原因就是Peter Marks本人钮药,他對Elevidys越是語焉不詳?shù)仡欁笥叶运瑢?shí)際批準(zhǔn)的可能性就越大诅订。在患者倡導(dǎo)組織CureDuchenne主辦的一次會(huì)議上氮栏,Peter Marks表示插棱,F(xiàn)DA的思維近年來發(fā)生了變化乡羹,變得更加關(guān)注患者利益。這種轉(zhuǎn)變促使FDA更加積極地尋找加速罕見病基因療法開發(fā)的方法肘何。Marks在會(huì)議上表示,“雖然FDA是一個(gè)監(jiān)管機(jī)構(gòu)艰膀,但法規(guī)最終必須服務(wù)于向患者提供產(chǎn)品這一基本控屡。因此,F(xiàn)DA正努力以患者為中心辫田,并利用這一點(diǎn)來協(xié)商法規(guī)峻维,以盡快實(shí)現(xiàn)這一目標(biāo)⊥┾”
Marks對罕見病基因療法的支持早已是公開的傾向,去年在另一個(gè)倡導(dǎo)團(tuán)體舉行的會(huì)議上刽肠,他主張靈活審查罕見病基因療法溃肪,同時(shí)駁斥有關(guān)加速審批的批評。如今Marks重申了他支持快速審批罕見病基因療法的態(tài)度音五。
最終的結(jié)果也的確不出意外惫撰,F(xiàn)DA在Elevidys的PDUFA日期前一天的6月20日將其加速批準(zhǔn)轉(zhuǎn)化為標(biāo)準(zhǔn)批準(zhǔn),并且擴(kuò)大了這款DMD藥物的患者年齡標(biāo)簽厨钻,將4-5歲患兒的適用范圍推廣到了所有年齡段扼雏,無論年齡大小或是否使用輪椅。需要指出的是莉撇,Elevidys的全面批準(zhǔn)適用于所有4歲及以上可以自行行走的患者呢蛤。對于依賴輪椅的患者,F(xiàn)DA授予了Elevidys加速批準(zhǔn)其障,仍然需要在更大規(guī)模的上市后驗(yàn)證性研究中確認(rèn)。對于攜帶幾種特定突變或已存在針對用于提供治療的病毒的抗體的男孩和男性恃感,該治療仍將無法使用斜曾。Sarepta 最近開始對已存在抗體的患者進(jìn)行研究。一項(xiàng)更大規(guī)模的驗(yàn)證性研究正在進(jìn)行中侦需,并將于秋季公布結(jié)果南椒。FDA在其聲明中稱坊蕴,“如果正在進(jìn)行的驗(yàn)證性試驗(yàn)結(jié)果為陰性呐取,F(xiàn)DA將不得不撤回加速批準(zhǔn)(針對輪椅患者)∩伲”
Elevidys獲批帶來的巨大爭議
對于像Elevidys這樣爭議很大的藥物,F(xiàn)DA的決定自然也招致了相當(dāng)大的質(zhì)疑聲音硅枷。有專家擔(dān)心其安全性旱婚,其它DMD基因療法試驗(yàn)中出現(xiàn)了患者死亡的事件,不過Elevidys并沒有出現(xiàn)類似的情況太迈。更為重要的是针执,如果未來幾年出現(xiàn)更好的基因治療或基因編輯治療围辙,接受Elevidys治療的患者可能無法接受那些療法我碟。幾乎所有此類治療都依賴于同一病毒家族將基因或基因編輯機(jī)制傳遞到肌肉細(xì)胞中。接受過一種治療的男孩隨后會(huì)產(chǎn)生抗體姚建,使他們無法接受另一種治療。
相對于Elevidys獲得全面批準(zhǔn)而言桥胞,這件事情在監(jiān)管尺度上引發(fā)的爭議似乎已經(jīng)超過了這款藥物本身的意義恳守,尤其是在未能通過上市后驗(yàn)證性III期研究的情況下仍然能夠得到監(jiān)管批準(zhǔn)并且擴(kuò)大標(biāo)簽范圍,F(xiàn)DA的監(jiān)管行為已經(jīng)在行業(yè)內(nèi)引發(fā)了很多爭議贩虾。
爭議的核心之一催烘,在于這個(gè)決定帶有很強(qiáng)的“一言堂“色彩沥阱。FDA發(fā)布的文件顯示,這個(gè)決定幾乎完全由CBER主任Peter Marks做出伊群。他推翻了三個(gè)審查小組和兩名高級副手的意見考杉。Marks的副手認(rèn)為,Sarepta提交的數(shù)據(jù)“對治療的好處存在很大的不確定性”舰始。然而Marks卻認(rèn)為崇棠,有必要加快對患有毀滅性罕見疾病的患者進(jìn)行基因治療。實(shí)際上惶芒,Elevidys去年獲得加速批準(zhǔn)的決定就是Marks本人的意志產(chǎn)生了主導(dǎo)性的作用燎匪。
對于FDA的批評者來說,Marks的這種行為過于“獨(dú)斷專行”赠搓,凸顯了人們對高管和患者權(quán)益倡導(dǎo)者(其中一些與行業(yè)有聯(lián)系)對決策者影響力過大的擔(dān)憂购畴,導(dǎo)致一些療效或安全性不足的藥物進(jìn)入市場验沮。在這一方面Elevidys并不是孤立铣才。FDA的高層官員之前還曾否決過監(jiān)管人員的決定,批準(zhǔn)了Sarepta的藥物Exondys 51(2016年)和Biogen的阿爾茨海默病藥物Aduhelm(2021 年)旱樊,這兩項(xiàng)批準(zhǔn)都引發(fā)了巨大的爭議。前FDA首席科學(xué)家Luciana Borio說:“我無法可說柑耙。Peter Marks嘲弄了幾十年來一直為患者服務(wù)的科學(xué)推理和審批標(biāo)準(zhǔn)述茂,這種行為也加劇了人們對FDA等科學(xué)機(jī)構(gòu)的不信任『螅” Luciana Borio曾在在2016年強(qiáng)烈反對Exondys的批準(zhǔn)朗涩。
FDA臨床評估辦公室主任Lola A. Fashoyin-Aje也在一封公開性中表達(dá)了類似的觀點(diǎn)绑改。信中指出,Eleviys在4至7歲兒童中連續(xù)兩次安慰劑對照試驗(yàn)均失敗兄一。沒有數(shù)據(jù)顯示患者在接受更多微肌營養(yǎng)不良蛋白治療后肌肉功能有所改善厘线。Sarepta僅提供了少數(shù)使用輪椅的患者的數(shù)據(jù),這些患者大多是12歲以上的杜氏肌營養(yǎng)不良癥男孩出革。盡管Sarepta出示的視頻表明Elevidys可能對某些患者有效造壮,但問題在于找出對于哪些類型的患者產(chǎn)生明確效果骂束。Lola A. Fashoyin-Aje和同僚建議Sarepeta進(jìn)行另一項(xiàng)臨床試驗(yàn)耳璧,但鑒于Marks的決定,這個(gè)建議目前看上去是不可行的展箱。
臨床和經(jīng)濟(jì)評論研究所 (ICER) 也對320萬美元的Elevidys的成本效益表示了質(zhì)疑蹬昌。他們在Elevidys獲批之前就在JAMA上的一篇文章中表示,“對于一項(xiàng)在2項(xiàng)隨機(jī)試驗(yàn)中未能達(dá)到主要終點(diǎn)且顯然不具有治愈效果的療法來說攀隔,320萬美元是一個(gè)巨大的代價(jià)”皂贩。
罕見病基因療法監(jiān)管的靈活性把握
FDA在加速批準(zhǔn)罕見病基因療法的監(jiān)管過程中所采用的靈活標(biāo)準(zhǔn)是一項(xiàng)備受爭議的措施,其支持者和反對者都有強(qiáng)烈的觀點(diǎn)嚎区。
支持者認(rèn)為拘挡,靈活標(biāo)準(zhǔn)對于罕見病患者是一個(gè)福音蝶桑。罕見病患者人數(shù)少掌社,傳統(tǒng)的大規(guī)模臨床試驗(yàn)難以進(jìn)行,靈活標(biāo)準(zhǔn)使得更多創(chuàng)新療法能夠更快地進(jìn)入市場窄切,為患者帶來新的治療希望。這些療法往往是這些患者唯一的治療選擇巍也,及時(shí)批準(zhǔn)可以挽救生命蜜硫,改善生活質(zhì)量。此外兵蟹,基因療法的科學(xué)基礎(chǔ)已經(jīng)有了長足的進(jìn)步谚碌,早期臨床試驗(yàn)數(shù)據(jù)雖然樣本量小,但也能提供足夠的有效性和安全性信息笆抱。靈活的審批流程還能激勵(lì)制藥公司加大對罕見病療法的研發(fā)投入,從而推動(dòng)整體醫(yī)療科技的進(jìn)步幼驶。
反對者則擔(dān)心艾杏,靈活標(biāo)準(zhǔn)可能帶來安全和有效性方面的隱患。由于罕見病患者基數(shù)小盅藻,早期臨床試驗(yàn)的數(shù)據(jù)可能不夠充分购桑,無法全面評估療法的長期效果和潛在風(fēng)險(xiǎn)。這種情況下氏淑,過早批準(zhǔn)可能會(huì)讓未充分驗(yàn)證的療法進(jìn)入市場勃蜘,給患者帶來意想不到的副作用或治療失敗的風(fēng)險(xiǎn)。此外假残,過于靈活的標(biāo)準(zhǔn)可能被一些企業(yè)利用缭贡,以快速商業(yè)化為目的,忽視療法的實(shí)際療效和安全性辉懒,從而損害患者利益阳惹。長期來看,這種做法可能削弱公眾對FDA監(jiān)管機(jī)制的信任,影響整體醫(yī)療體系的公信力琢播。
總的來說爹故,F(xiàn)DA在加速批準(zhǔn)罕見病基因療法過程中所采用的靈活標(biāo)準(zhǔn),既有助于滿足迫切的醫(yī)療需求沥院,也存在潛在的風(fēng)險(xiǎn)和挑戰(zhàn)妹茬。如何在加快創(chuàng)新療法上市與保障患者安全之間找到平衡篇胰,是監(jiān)管機(jī)構(gòu)需要持續(xù)面對和解決的關(guān)鍵問題泳唇。
FDA認(rèn)識(shí)到罕見病具有高度多樣性,患病率殃描、進(jìn)展速度、臨床表現(xiàn)和異質(zhì)性程度各不相同扒哩。因此聚伤,雖然所有試驗(yàn)都必須遵循嚴(yán)格的質(zhì)量和藥物警戒協(xié)議,但許多行業(yè)領(lǐng)導(dǎo)者主張?jiān)跐M足監(jiān)管標(biāo)準(zhǔn)方面具有更大的靈活性虱怖。
例如坠天,Peter Marks最近主張?jiān)跍y試罕見病基因療法時(shí)進(jìn)行非隨機(jī)單臂試驗(yàn),支持生物標(biāo)志物和替代終點(diǎn)來支持批準(zhǔn)座咆。2023年,美國政府還啟動(dòng)了支持臨床試驗(yàn)推進(jìn)罕見病治療 (START) 試點(diǎn)計(jì)劃介陶,該計(jì)劃使贊助商能夠更頻繁地與FDA工作人員溝通堤舒,并提供解決臨床開發(fā)問題的機(jī)制。這些舉措的目標(biāo)就是縮短開發(fā)時(shí)間哺呜,讓研究人員和患者都更容易獲得罕見病研究舌缤。然而,并不存在一種通用的解決方案可以有效地將療法帶給患者某残。
監(jiān)管機(jī)構(gòu)可能認(rèn)為單臂試驗(yàn)不適合非常異質(zhì)的疾病人群或太小的樣本組,罕見病藥物仍必須有大量證據(jù)表明其有效性和安全性才能獲得FDA批準(zhǔn)驾锰。因此卸留,生物制藥公司必須考慮每種疾病和藥物研究的具體參數(shù)走越,使試驗(yàn)設(shè)計(jì)與監(jiān)管標(biāo)準(zhǔn)保持一致椭豫。
FDA還鼓勵(lì)贊助商評估生物標(biāo)志物的識(shí)別和使用。研究人員必須收集足夠的信息來支持每個(gè)生物標(biāo)志物并驗(yàn)證檢測方法旨指,這一領(lǐng)域更多的靈活性正在推動(dòng)療法開發(fā)和試驗(yàn)設(shè)計(jì)的進(jìn)步宛殉。此外,許多生物制藥公司正在開展更多以患者為中心的研究希镶,以解決小群體參與者的問題擂益。
總體而言,F(xiàn)DA在監(jiān)管罕見病療法開發(fā)的過程中展示了顯著的靈活性翻妆。這種靈活性體現(xiàn)在加速審批通道的建立、臨床試驗(yàn)設(shè)計(jì)的靈活性欧纬、替代終點(diǎn)的使用以及患者和利益相關(guān)者的參與等方面践拐。這些措施不僅促進(jìn)了罕見病療法的創(chuàng)新和研發(fā),也大大加速了新療法的上市祭啸,從而滿足了罕見病患者的迫切需求鹿逞。未來,隨著科技的進(jìn)步和對罕見病理解的加深俐逛,F(xiàn)DA可能會(huì)繼續(xù)優(yōu)化和完善這些靈活性措施,以更好地支持罕見病療法的開發(fā)和審批桑嘶。
編者按:
Viltepso和Elevidys兩款藥物的審批歷程揭示了監(jiān)管機(jī)構(gòu)在平衡創(chuàng)新與患者安全方面的挑戰(zhàn)炊汹。FDA在加速批準(zhǔn)罕見病基因療法方面采取了靈活標(biāo)準(zhǔn),旨在為患者提供新的治療選擇逃顶。然而兵扬,這種靈活性也引發(fā)了關(guān)于安全性和有效性的擔(dān)憂。展望未來口蝠,如何在加快創(chuàng)新療法上市與保障患者安全之間找到最佳平衡點(diǎn),將是FDA未來需要持續(xù)關(guān)注的關(guān)鍵問題妙蔗。
參考文獻(xiàn)
Mast, J. Duchenne muscular dystrophy drug from Nippon Shinyaku fails in rare confirmatory trial. STAT. 27. 05. 2024.
Fidler, B. With Duchenne decision ahead, FDA’s Marks pushes for speedy gene therapy approvals. Biopharma Dive. 24. 05. 2024.
Mast, J. FDA approves Sarepta’s Duchenne gene therapy for nearly all patients. STAT. 20. 06. 2024.
Mast, J. et al. Top FDA official Peter Marks overruled staff, review team to approve Sarepta gene therapy. STAT. 20. 06. 2024.
Keshavan, M. ICER casts doubt on Sarepta’s DMD gene therapy. STAT. 03. 05. 2024.
 產(chǎn)業(yè)資訊
深藍(lán)觀 2024-11-28
13
產(chǎn)業(yè)資訊
深藍(lán)觀 2024-11-28
13
 產(chǎn)業(yè)資訊
瞪羚社 2024-11-28
17
產(chǎn)業(yè)資訊
瞪羚社 2024-11-28
17
 產(chǎn)業(yè)資訊
丹諾醫(yī)藥 2024-11-28
15
產(chǎn)業(yè)資訊
丹諾醫(yī)藥 2024-11-28
15
 熱門資訊
熱門資訊 微信公眾號(hào)
微信公眾號(hào) 熱點(diǎn)標(biāo)簽
熱點(diǎn)標(biāo)簽